Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Virol Antivir Res Vol: 110 Issue: 4
Virtual Screening Reveals Potential Anti-Parasitic Drugs Inhibiting the Receptor Binding Domain of SARS-CoV-2 Spike protein
Sathya Muthusamy, Hariprabu Gopal, Thiliban Manivarma, Satya Narayan Pradhan, Prince. R. Prabhu*
Department of Biotechnology, Anna University, Chennai--600025, India
- Corresponding Author:
- Prince. R. Prabhu
Department of Biotechnology
Anna University, Chennai--600025, India
Phone: +91 9840924463
E-mail: princeprp@gmail.com
Received: June 13, 2021 Accepted: July 08, 2021 Published: July 08, 2021
Citation: Muthusamy S, Gopal H, Manivarma T, Pradhan SN, Prabhu PR (2021) Virtual Screening Reveals Potential Anti-Parasitic Drugs Inhibiting the Receptor Binding Domain of SARS-CoV-2 Spike protein. J Virol Antivir Res 10:4.
Abstract
Abstract The 2019’s COVID-19 outbreak which spread to over 200 countries across the globe had its origin from the 2002’s SARS-CoV-1 epidemic. The corona viruses are single stranded positive sense RNA viruses with 4 structural proteins such as spike(S), membrane(M), envelope(E) and nucleocapsid(N) proteins and 16 non-structural proteins (NSPs). The spike(S) protein is a homo-trimer protruding from the viral surface comprising 2 subunits namely, the S1 and S2 where the S1 subunit consists of the receptor binding domain (RBD) and the S2 subunit consists of the fusion peptide. The spike glycoprotein is considered as the most desired pharmacological target for drug designing, thus blocking the viral entry into the host. Computer-Aided Drug Designing significantly reduces the cost and time in drug discovery compared to the in-vitro methods. Hence in our study, we have performed a virtual screening of the complete set of anti-parasitic drugs using the popular molecular docking tool, Autodock vina with an aim to repurpose the potential hits for the SARS-CoV-2 infection. The repurposed drugs are advantageous for their easy and immediate access owing to their already proven safety records in toxicity and hence are better than novel drugs. Our results revealed 32 anti-parasitic compounds crossing our threshold binding affinity with selamectin, ivermectin, artefenomel, moxidectin, posaconazole, imidocarb, piperaquine, cepharantine, betulinic acid and atovaquone at the top of the list and occupying the three different electrostatic regions in the RBD. Further optimization strategies and in-vitro trials could make our potential anti-parasitic hits, a potential cure for the SARS-CoV-2 infection. Keywords Virtual screening; SARS-CoV-2; COVID-19; Spike protein; Receptor Binding Domain; Autodock Vina, Antiparasitic drugs; Anti-viral; Repurposed drugs. Abbreviations COVID-19: COrona VIrus Disease 2019; SARS-CoV-1: Severe Acute Respiratory Syndrome-Corona Virus-1; SARS-CoV-2: Severe Acute Respiratory Syndrome-Corona Virus-2; S protein: Spike protein; RBD:Receptor Binding Domain; ACE-2: Angiotensin Converting Enzyme-2; NSP: Non Structural Protein; RNA: Ribo Nucleic Acid; CDC: Centre for Disease Control and Prevention; WHO: World Health Organization; RT-PCR: Reverse Transcription.
Keywords: Virtual screening; SARS-CoV-2; COVID-19; Spike protein; Receptor Binding Domain; Autodock Vina, Antiparasitic drugs; Anti-viral; Repurposed drugs.
Keywords
Virtual screening; SARS-CoV-2; COVID-19; Spike protein; Receptor Binding Domain; Autodock Vina, Antiparasitic drugs; Anti-viral; Repurposed drugs.
Abbreviations
COVID-19: COrona VIrus Disease 2019; SARS-CoV-1: Severe Acute Respiratory Syndrome-Corona Virus-1; SARS-CoV-2: Severe Acute Respiratory Syndrome-Corona Virus-2; S protein: Spike protein; RBD:Receptor Binding Domain; ACE-2: Angiotensin Converting Enzyme-2; NSP: Non Structural Protein; RNA: Ribo Nucleic Acid; CDC: Centre for Disease Control and Prevention; WHO: World Health Organization; RT-PCR: Reverse Transcription Polymerase Chain Reaction; GISAID: Global Initiative on Sharing All Influenza Data; HR-1: Heptad Repeat 1; HR-2: Heptad Repeat 2; CADD: Computer-Aided Drug Design; FDA: Food and Drug Administration; GABA: Gamma-Amino Butyric Acid; CNS: Central Nervous System; BBB: Blood Brain Barrier; CASF: Comparative Assessment of Scoring Function; PDB: Protein Data Bank; SDF: Structure Data File; cryo-EM: cryogenic Electron Microscopy; RCSB: Research Collaboratory for Structural Bioinformatics; C-I-TASSER: Contact-guided Iterative Threading ASSembly Refinement; RMSD: Root Mean Square Deviation; RdRp: RNA dependent RNA Polymerase; 3CLpro: 3 Chymotrypsin Like protease; Mpro: Main protease; PLpro: Papain Like protease; NTZ: Nitazoxanide; HA: Hydrogen bond Acceptors; HD: Hydrogen bond Donors; HIV: Human Immunodeficiency Virus
Introduction
The COVID-19 pandemic startling the humankind across the globe marked its emergence from the SARS epidemic of 2002. The 2002’s SARS-CoV-1 emerged from the Guangdong province of China and culminated with the infection of 8000 people and causing 774 deaths [1]. The recent SARS-CoV-2 infection was discerned following a sudden outbreak of an atypical pneumonia with unknown disease etiology in the Wuhan city of China during the late December 2019 [1, 2]. The ceaseless efforts of WHO in association with the Centre for Disease Control and Prevention (CDC) to decipher the disease pathogen paid of its results on January 10, 2020 when the first whole genomic sequence of 2019-nCoV was released which helped the researchers to quickly identify the virus in the patients using reverse transcription- polymerase chain reaction (RT-PCR) method [2, 3]. The chronology of the pandemic progress has been in par with interventions to curb the same. The major mile stone starts from 21st January 2020, when the first paper was published with evidences to prove that the 2019nCoV shares ancestry with bat corona virus HKU9-1 and is similar to the SARS-CoV-1 of 2002. By 31st January 2020, 51 whole genome sequences were deposited in the GISAID database and on 12th February, the WHO renamed the 2019-nCoV as SARS-CoV-2 and the disease as COVID-19. On 11th March 2020, the WHO recognized COVID-19 as a global pandemic which then reached a global death toll of 9913 and about 2,42,650 laboratory confirmed cases by 19th March 2020. Thus the global fatality rate was 3.92% [3]. The outbreak spread to over 200 countries including Italy, Iran, France, Spain, Germany, UK, USA and India with more than 3.2 million confirmed cases, 0.23 million confirmed deaths and 10 million recoveries as of the statistics of 1st May, 2020 [2,4].
The corona viruses are enveloped single stranded positive sense RNA viruses with the unique feature of a “club-like” projection from the vision surface which mimics a crown and hence the name. The corona viruses being the largest RNA viruses known so far are about 32kb large, which encodes for 4 structural and 16 non-structural proteins (NSPs). The non-structural proteins aid in modulating the innate immunity by suppressing the interferon synthesis and thereby regulating its signalling. While the non-structural proteins occupy 2/3rd (20kb) of the viral genome, the remaining 12 kb is occupied by the structural proteins namely spike(S), membrane (M), envelope (E) and nucleocapsid (N) proteins. The S, M, and E proteins are membrane bound, while the N protein is located within the virions in complex with the genomic RNA [5]. The S protein forms a homo-trimmer protruding from the viral surface and is highly immunogenic to the host immune system. The S protein has 2 functional subunits such as the S1 subunit containing the receptor binding domain(RBD) which directly interacts with the human ACE-2(Angiotensin Converting Enzyme-2) receptor and the S2 subunit containing the fusion peptide which aids in membrane fusion [1,6]. The spike proteins of SARSCoV- 1 and SARS-CoV-2 display high structural homology and conserved ectodomains with 89.8% sequence similarity. However, the binding affinity of the S protein with the human ACE-2 receptor is 10 to 20 fold higher for SARS-CoV-2 than it is for SARS-CoV-1 [7]. An effective therapeutic strategy for preventing the viral infection could be to create an interference with the viral entry [6]. Thus the host cell receptor ACE-2, an exopeptidase found across the epithelial cells of the respiratory tract, is identified as the most suitable pharmacological target for blocking the interaction of S protein with the host [4]. The mechanism of infection sets about with the binding of the receptor binding domain(RBD) in the S1 subunit of the S protein to the human ACE-2 receptor followed by the interaction of heptad repeat-1(HR-1) and heptad repeat- 2(HR-2) domains in the S-2 subunit of S protein. This interaction subsequently results in the formation of a six helix bundle (6HB) fusion core which brings the viral and cellular membranes into close proximity for fusion and infection [7].
The prevailing economic crisis and daunting situation across the globe urges the medical and scientific community to arrive at a potential cure for the pandemic at the earliest. Regrettably, the process of drug discovery is an iterative process which, depending on the strategy used, comprises several discrete stages such as, target identification and validation; assay development; screening(whole cell or molecular target based) to identify hits; procurement/synthesis and assessment of analogues to develop structure-activity relationships(SAR) and identify leads [8]. This long and tedious process to hunt a lead molecule often demoralizes the researchers by the endless possibilities one has to search through [9]. The lead identification is the followed by iterative medicinal chemistry to optimize leads; and preclinical development prior to clinical evaluation [8]. The superseding preclinical and clinical trial phases test for a variety of toxicities and adverse drug effects since safety is always the most important issue during drug development [10].
Fortunately, the computational tools available to predict the protein-ligand binding have come to the rescue and have offered a valuable help in rationalizing the path to drug discovery [9, 11]. Virtual screening is nowadays a standard step before wet-lab experiments that continue to show promise in hit identification and subsequent optimization [12, 13]. The CADD (Computer Aided Drug Design) approach significantly reduces the cost and time compared to the trial-and-error method using experimental studies [14, 15]. Among the wide range of computational tools, molecular docking which uses a structure-based computational strategy to predict the binding efficiency between the drug and target molecule has swiftly gained ranks to secure a valuable position in the modern scenario of structure based drug design [9, 16, 17].
The discovery of a novel drug molecule and its development is a long journey full of high risks wherein, converting the original synthetic molecule to a metabolically robust, orally bioavailable drug can be extremely challenging with very long time lines [8, 10]. In recent years the average cost of developing a new drug reaches up to 2.6 billion U.S dollars and the estimated attrition rate of drug candidates is up to 96%. This high attrition rate is due to the drug’s safety which accounts for 30% of the total drug failures. Even after the approval of a drug in the market it could be withdrawn during pharmacovigilance, owing to its adverse side-effects [10]. Therefore, a rational strategy to treat COVID-19 is the repurposing of already FDA- approved drugs with proven safety records [2, 18].
Here in this study, we have virtually screened the predominant anti-parasitic agents available in the Drug Bank and Pub hem databases against the spike protein of SARS-CoV-2 for the fact that these antiparasitic drugs rarely penetrate the blood-brain barrier in the CNS and hence act along a defended safety line of action when compared to other drugs [19]. The anti-parasitic drugs work by potentiating glutamate-gated chloride ion channels and GABA gated channels in the parasites. Subsequent neurotoxicity, paralysis and death of the parasites are caused by increased permeability to chloride ions and hyperpolarization of nerve cells. Since mammals including humans lack the glutamate gated chloride channels, and that these drugs do not cross the blood-brain barrier (BBB) in the CNS of mammals, these anti-parasitic drugs do not produce adverse effects when administered in humans. Also, these anti-parasitic drugs produce longer and more sustained plasma concentrations [19]. The results of our virtual screening showed several substantial hits having appreciable binding affinities to the RBD of the spike protein with selamectin, Ivermectin, Artefenomel, Moxidectin, Posaconazole, Imidocarb, Piperaquine, Cepharantine, Betulinic acid and Atovaquone topping our list. We also found that the inhibitory concentration of our top-ranked ant parasitic drugs against the spike protein was in micromolar range, suggesting that the toxicity with humans as comparatively less to the other drugs.
Materials and Methods
Docking software
We performed molecular docking using the software Auto dock Vina version 1.1.2 [20]. Auto dock Vina was developed to improve the accuracy of binding predictions of its predecessor Autodock4. It was improvised to achieve further speed-up from parallelism, by using multi-threading on multicore machines [20]. All the docking software’s are generally composed of two components, namely the scoring function which estimates the free energy of the modelled system and the search algorithm or exploration method used to sample the positional and conformational space [11]. The search algorithm used in the Vina program is based on local optimization and the scoring function is knowledge-based and fully empirical(hybrid scoring function), including Gaussian steric interaction terms, a finite repulsion term, piecewise linear hydrophobic and hydrogen bond interaction terms and an entropic term proportional to the number of rotatable bonds [11,13,14,21]. Auto dock Vina was the best of all methods in terms of docking power at the CASF-2013 benchmark. The CASF (Comparative Assessment of Scoring Functions) is a benchmark of scoring functions which evaluates the strengths and weaknesses of various docking programs with four criteria such as scoring power, ranking power, docking power and screening power [11]. The Vina program is up to two orders of magnitude faster than Autodock4 while also significantly improving the accuracy of binding mode predictions [13, 20]. Its multicore capacity, high performance and enhanced accuracy with ease of use and free availability have contributed to an extremely fast dissemination through the docking community [13].
Data collection
Our list of ligands which had the complete set of all antiparasitic drugs were retrieved from the Drug Bank [22] and PubChem [23] chemical libraries. The 3D structures of the ligands were downloaded in SDF format from the databases and converted to individual PDB files using Open Babel version 2.4.1 [24]. In order to perform protein structural analysis, the full length amino acid sequence of SARS-CoV-2 spike protein was obtained from the GenBank database(GenBank ID: QHD43416) [25] and two protein structures were retrieved from the RCSB Protein Data Bank(PDB) [26] which were the cryo- EM structure of spike protein in open state(PDB ID:6VYB) with a resolution of 3.2Aº(Figure 1a) and the crystallographic structure of spike protein’s receptor binding domain(RBD)in complex with the ACE-2 receptor(PDB ID:6LZG) with a resolution of 2.5Aº(Figure 1b). To conduct docking, the structural model of the full length SARS-CoV-2 spike protein (Figure 2a) was obtained through C-ITASSER( Contact-guided Iterative Threading ASSEmbly Refinement) , also known as the “Zhang server”of the University of Michigan, USA [27] which is a top ranked automated server for protein structure prediction [28].

Figure 1: Structure of SARS-CoV-2 spike protein (A) A cartoon representation of the crystal structure of the RBD in complex with the ACE-2 receptor.(PDB ID: 6LZG). (B) A cartoon representation of the superimposed cryo-EM structure of the spike protein’s chain A(PDB ID: 6VYB)(in red) on the crystal structure of RBDACE- 2 receptor complex(PDB ID:6LZG)(in green). (C) A cartoon representation of chain A in the cryo-EM structure of spike protein(PDB ID:6VYB).

Figure 2: C-I-TASSER protein model (A) A cartoon representation of the computationally solved structure of the spike protein by C-I-TASSER.
(B) A cartoon superposition of C-I-TASSER’s protein model(in magenta) on the cryo-EM structure of spike protein(PDB ID: 6VYB)(in green).
Structural analysis of protein
The chain A in the cryo-EM spike protein structure (PDB ID: 6VYB) was superimposed on the complex crystal structure of RBD with spike protein (PDB ID: 6LZG) using the Pymol molecular visualization tool [29] to analyze and visualize the region of RBD on the spike protein. The superposition disclosed the range of amino acids in the RBD of spike protein which started at position ASN331 and ended at position VAL524 encompassing a total of 193 residues in the entire sequence. This information later helped us fix the central atom co-ordinates and the search space for docking (Figure 1c superimposed structure).
The experimental structure of the spike protein (6VYB) was found to have more than 200 missing residues in its entire chain length. The cryo-EM structure covered only75% of the total residues with several residues on the receptor binding domain (RBD) missing, which made it unsuitable for docking studies [28]. Therefore, we used the computationally modeled spike protein obtained from C-ITASSER[ 27,30-33] for docking which is available at https://zhanglab. ccmb.med.umich.edu/COVID-19/ .C-I-TASSER is an extended pipeline of I-TASSER and utilizes deep convolutional neural network based contact maps to guide the Monte Carlo fragment assembly simulations[27,30,31-39]. When the computational model of C-ITASSER was superimposed on the experimental spike protein structure, the C-I-TASSER model shared a high structural similarity with an estimated TM score of 0.84 to the cryo-EM structure (Figure 2b).
Protein and ligand preparation
The preparation of protein target for the molecular docking involves removal of the water molecules and native ligands attached with target and other hetero-atoms, which may provide hindrance while docking. Thus the rigid receptor and the flexible ligands were suitably parameterized for docking via auto dock Tools 1.5.6 [40] after adding polar hydrogen’s and Gasteiger charges. The parameterized systems were recorded in the PDBQT files. PDBQT format is an extension of the PDB format with additional fields (partial charge’Q’ and atom type’T’) for ATOM and HETATM records and information about the rigid blocks of molecules [11].
Docking parameters
The interaction grids were generated using the configuration file. The grid was positioned at the C atom of the residue LEU425 with the grid co-ordinates as center_x = -12.605, center y = -9.290 and center_z = 518.051. The grid dimensions were chosen so as to include the complete receptor binding domain (RBD) in the target and thus augmented by 50Aº in ±x, ±y and ±z directions. The default exhaustiveness value of 8 was employed, which controls the number of times to repeat the calculations within the search space [12]. Unlike Autodock4, theVina program automatically calculates the grid maps and clusters in the results [20].
Docking and enrichment strategy
Docking was performed with the auto dock Vina program version 1.1.2 [20]. In molecular docking, the best conformers are represented with the lowest binding energy (-kcal/mol) and a pose is considered native if its RMSD (non-hydrogen atoms) with respect to the experimental ligand pose is ≤2 Aº [11]. Therefore, our enrichment strategy to select the ligands was fixed with a threshold score of binding energy as -7 kcal/mol whose native poses with the least RMSD were considered for post-docking analysis.
Results
Our results had 32 potential anti-parasitic candidates crossing the threshold score of binding energy and effectively inhibiting the RBD of spike protein by forming a number of non-bonded interactions with its residues. The top 10 anti-parasitic inhibitors are mentioned in the Table 1 namely, selamectin, ivermectin, artefenomel, moxidectin, posaconazole, imidocarb, piperaquine, cepharantine, betulinic acid and atovaquone with binding affinities ranging from -7.7 kcal/mol to -11.2 kcal/mol.
Table 1: List of top ranked antiparasitic drugs: A tabular column representation of the putative anti-parasitic compounds which were found to act as effective anti-virals against the spike protein of coronavirus along with their molecular and druggable properties.
| S.No | Drug | Pubchem ID | Molecular formula | Structural formula | Binding affinity (kcal/mol) | Total polar surface area(TPSA) | Hydrogen bond acceptors(HA) | Hydrogen Bond donors(HD) | Molecular weight(g/mol) | Number of Lipinski rule violations |
|---|---|---|---|---|---|---|---|---|---|---|
| 1. | Selamectin | CID9578507 | C43H63NO11 | 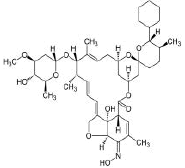 |
-11.2 | 154.73 | 12 | 3 | 769.96 | 2 (MW>500, NorO>10) |
| 2. | Ivermectin | CID6321424 | C48H74O14 | 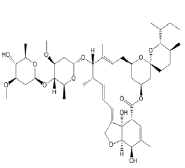 |
-9.3 | 170.06 | 14 | 3 | 875.09 | 2 (MW>500, N or O>10) |
| 3. | Artefenomel | CID24999143 | C28H39NO5 | 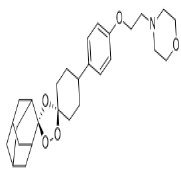 |
-8.5 | 49.39 | 6 | 0 | 469.61 | 0 |
| 4. | Moxidectin | CID9832912 | C37H53NO8 | 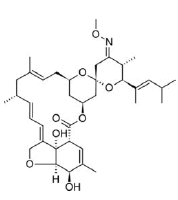 |
-8.5 | 116.04 | 9 | 2 | 639.82 | 1(MW>500) |
| 5. | Posaconazole | CID468595 | C37H42F2N8O4 | 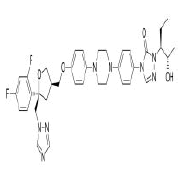 |
-8.3 | 115.7 | 9 | 1 | 700.78 | 3(MW>500, N or O>10, NH or OH>5) |
| 6. | Imidocarb | CID21389 | C19H20N6O | 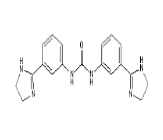 |
-8 | 89.91 | 3 | 4 | 348.4 | 0 |
| 7. | Piperaquine | CID122262 | C29H32Cl2N6 | 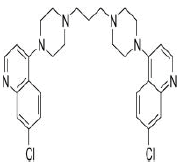 |
-7.9 | 38.74 | 4 | 0 | 535.51 | 1(MW>500) |
| 8. | Cepharantine | CID7098680 | C37H38N2O6 | 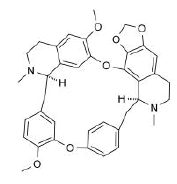 |
-7.9 | 61.86 | 8 | 0 | 606.71 | 1(MW>500) |
| 9. | Betulinic acid | CID64971 | C30H48O3 | 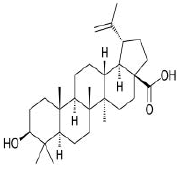 |
-7.9 | 57.53 | 3 | 2 | 456.7 | 1(MLOGP>4.15) |
| 10. | Atovaquone | CID74989 | C22H19ClO3 | 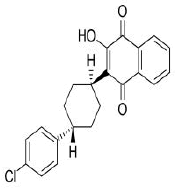 |
-7.7 | 54.37 | 3 | 1 | 366.84 | 0 |
In a study conducted by Sruthi. Unni et al [1], the receptor binding domain(RBD) was found to have three important sites based on the electrostatic surface region at the S protein-ACE-2 interface and hence was divided into three regions namely site 1, site 2 and site3(Figure 3a). The hydrophilic ‘site 1’ consists of residues GLY446, TYR449, GLY496, GLN498, THR500 and ASN501 on the S protein interacting with the residues ASP38, TYR41, GLN42, LYS353 and ASP355 on the ACE-2 receptor surface. The moderate hydrophilic ‘site 2’ region comprised of residues LYS417 and GLN493 in the RBD interacting with the residues ASP30 and GLU35 on the ACE-2 receptor. The ‘site 3’ which was also a moderate hydrophilic region consisted of residues ALA475 and ASN 487 in the RBD interacting with the residues SER19, GLU24 and TYR83 of the ACE-2 receptor. Further detailed analysis indicated that the topology of the binding site is a substantial hydrophilic region on the head end of the ‘site 1’ region. The ‘site 1’ also houses a very prominent hydrophobic cleft produced by the residues TYR495, PHE497 and TYR505. Though this cleft does not indulge in interactions with the ACE-2 receptor surface, it seems to be an implementable strategy while designing drugs for the S protein binding to inhibit the receptor interaction. The cleft is followed by the ‘site 2’ residues, which provide a good hook position for the potential drug compounds. There are also many hydrophilic non-interacting residues like ARG403, GLU406, TYR453 in the ‘site 2’ region which again provide a potential strategy to interrupt the S protein-ACE-2 interaction [1].Therefore, the 32 potential inhibitors were classified under the three sites as mentioned in the Table 2, based on the closest proximity they bind to in the RBD, which would help building a further optimization strategy of the inhibitors. In addition, the nature of charges present on the protein surface was also found (Figure 3b) using the Pymol molecular visualization tool in order to get a clear picture about the electrostatic properties of the spike protein’s RBD.

Figure 3: Electrostatic surfaces on the receptor binding domain(RBD). (A) A cartoon representation of the receptor binding domain with the hydrophilic ‘site 1’ in yellow; moderate hydrophilic ‘site 2’ in green; moderate hydrophilic ‘site 3’ in red; hydrophobic cleft of the ‘site 1’ in magenta and the non-interacting hydrophilic residues of the ‘site 2’ in cyan. (B) A surface representation of the receptor binding domain(RBD) depicting the positively charged surfaces in blue, negatively charged surfaces in red and neutral(uncharged) surfaces in white.
Table 2: Classification of drugs according to the electrostatic: A tabular representation of the putative drugs classified based on the various electrostatic regions they bind to in the RBD of the spike protein along with the information of the interacting residues and the types of non-bonded interactions in the vicinity.
| S.No | Region in RBD | Drugs | Interacting residues | Types of non-bonded interactions |
|---|---|---|---|---|
| 1. | Site 1 | Atovaquone | SER514, GLU516, TYR396, PHE464 | h bond(1), hydrophobic(4) |
| Praziquantel | ARG355, PHE515, PHE429, TYR396, LEU518 | h bonds(3), hydrophobic(4) | ||
| Artemisnin | LEU517, LEU518, GLU516, PHE464 | h bonds(3), hydrophobic(1) | ||
| Mefloquine | PHE515, SER514, GLU516, TYR396, PHE429, LEU518 | h bonds(2), halogen(4), electrostatic(1), hydrophobic(7) | ||
| Tetrandrine | LEU517, GLU516, PHE429, LEU518 | h bonds(3), hydrophobic(6) | ||
| Flubendazole | ARG355,ARG466,LEU517,GLU516, PHE464, PHE515, PHE429 | h bonds(5), halogen(3), electrostatic(2), hydrophobic(1) | ||
| 2. | Site 2 | Ivermectin | GLU340, ARG466, PRO337, ARG355, ARG357, TYR396 | h bonds(3), electrostatic(3), hydrophobic(3) |
| Moxidectin | LEU455, LYS417 | hydrophobic(2) | ||
| Posaconazole | TYR421, PHE456, TYR489 | h bond(1), electrostatic(2), hydrophobic(2) | ||
| Cepharantine | PHE515, ARG355,GLU465, TYR396, PHE429 | h bonds(2), electrostatic(2), hydrophobic(3) | ||
| Betulinic acid | PHE429 | hydrophobic(1) | ||
| Phenothrin | TYR396, PHE429, PHE464, LEU518 | hydrophobic(6) | ||
| Pyronaridine | GLU465, PRO426, PRO463, PHE429, PHE464, PRO426 | electrostatic(2), hydrophobic(6) | ||
| Dithiazine | ARG466, PHE429, PHE464, ILE468 | h bond(1), electrostatic(2), hydrophobic(4) | ||
| Radicicol | TYR396, PHE429 | hydrophobic(3) | ||
| 3. | Site 3 | Selamectin | GLU465, PHE464 | h bonds(2), electrostatic(2), hydrophobic(3) |
| Artefenomel | ARG466, GLU465, TYR396, PHE429, PHE464 | h bonds(3), electrostatic(1), hydrophobic(4) | ||
| Imidocarb | GLN474, TYR489, GLU471, LYS458, ILE472, THR478, TYR473, ALA475 | h bonds(6), hydrophobic(3) | ||
| Piperaquine | LYS458, ALA475, ARG457, TYR421, PHE456, ILE418 | h bonds(4), hydrophobhic(7) | ||
| Pyrvinium | GLU465, ARG466, TYR396, PHE429, ILE468 | h bonds(2), electrostatic(1), hydrophobic(4) | ||
| Mebendazole | ARG355, ARG466, GLU465, PHE464, PHE429 | h bonds(7), electrostatic(2), hydrophobic(2) | ||
| Diclazuril | ARG355, ARG466, GLU465, TYR396, PHE429, PHE464 | h bonds(3), electrostatic(2), hydrophobic(5) | ||
| Artesunate | ARG355, ARG466, SER514 | h bonds(3) | ||
| Monensin | TYR489, PHE490 | h bonds(2) | ||
| Mefloquine | TYR489, LYS458, SER459, PHE456, ALA475, ARG457, TYR421 | h bond(1), halogen(3), hydrophobic(5) | ||
| Puromycin | ARG457, GLN474, SER477, LEU455, TYR473, PHE456, ILE418, TYR421, LYS458 | h bonds(5), hydrophobic(7) | ||
| Sulfaquinoxaline | ARG457, SER477, LYS458, SER459 | h bonds(4), hydrophobic(3) | ||
| Emetine | LYS458, SER459, GLN474, ILE418, LYS458, PHE456, TYR473, ALA475 | h bonds(3), hydrophobic(6) | ||
| Oxfendazole | ARG457, GLN474, LYS458, SER459 | h bonds(2), hydrophobic(3) | ||
| Niclosamide | TYR489, GLN474, SER477, PRO479, GLU471, CYS480, LYS458, SER459 | h bonds(5), electrostatic(1), hydrophobic(2) | ||
| Fenbendazole | ARG457, GLN474, TYR421, LYS458, SER459 | h bonds(2), hydrophobic(4) | ||
| Artenimol | ARG457, TYR489, PHE456 | h bonds(2), hydrophobic(1) |
The entire workflow of the protein structural analysis and the virtual screening is summarized in Figure 4.

Figure 4: Flowchart depicting the workflow of protein structural analysis and virtual screening.
Discussion
The spike protein target and its RBD
Previous research efforts to develop antiviral agents against the members of Coronaviridae family demonstrated a number of suitable drug targets such as, the RNA dependent RNA polymerase (RdRp), 3-Chymotrypsin like protease (3CLpro) also called as the main protease (Mpro), Papain like protease (PLpro) and the spike protein-ACE-2(Angiotensin converting enzyme-2) entry receptor aiming to block the critical corona virus host infections [41]. Of the several proteins that can be potential druggable targets in SARSCoV- 2, the receptor binding domain(RBD) of the spike protein is one of the most desired molecular targets because of its cardinal role in host cell binding and also, the spike protein’s major receptor for both SARS-CoV-1 and SARS-CoV-2 is the human Angiotensin Converting Enzyme(ACE-2) [5,42]. The SARS-CoV-2 expresses a cell surface spike(S) protein in which a specific segment called the receptor binding domain(RBD) plays a vital role in infecting the host by binding to the ACE-2 receptor in the host [42]. The spike glycoprotein which is a class-I fusion protein plays a seminal role in the viral infection, starting from being the major driving force for host cell recognition; mediating the fusogenic mechanism of the viral envelope with the host cells, where the receptor recognition and attachment of S protein to the ACE-2 receptor is a pre-requisite step and key determinant of the host cell and tissue tropism [43-45]. The glycosylated spike protein is responsible for the primary establishment of the host-protein direct interaction through its RBD, which binds to the ACE-2 receptor on the host cell plasma membrane [1, 16]. The ACE-2 is a host cell exopeptidase and metallocarboxypeptidase that catalyses the conversion of angiotensin-1 to the nonapeptide angiotensin and the conversion of angiotensin-II to angiotensin 1-7, to initiate S protein mediated cell entry [4]. Thus the spike protein of corona viruses is a distinguishing feature that is the basis not only for its name, but also the myriad roles in facilitating the host cell attachment and entry [5]. Recent researches speculated that the residues from 331 to 524 which constitute the receptor binding domain (RBD) is the most crucial target for computational docking [46]. There are also additional evidences obtained from in-silicon structural modelling about the RBD which illustrate that the interface segment of RBD might be acquired by SARS-CoV-2 via a complex evolutionary process rather than mutation accumulation [47]. Also several important residues have been found to maintain the stability of the interface during the S protein-ACE-2 binding process such as GLN493, PRO499, GLN403, LYS451 and ASP416 [15, 47]. Therefore, the ACE-2 receptor constitutes a molecular target to inhibit the cell entry of SARS-CoV-2 and the S protein can be considered as a first line of therapeutic target for antiviral therapy and vaccine development [1, 4].
Repurposed drugs
There has been several repurposed drugs against SARS-CoV-2 reported in various literature sources irrespective of their target protein such as chloroquine phosphate, hydroxychloroquine, lopinavir, ritonavir, umifenovir, remdesivir, favipravir, atazonavir, ribavirin, tocilizumab and meplazumab(monoclonal antibodies), cepharantine, selamectin, mefloquine hydrochloride, nitazoxanide and ivermectin [2,4,18,48,49]. While considering only the spike protein target, there is convincing evidence that chloroquine, hydroxychloroquine, selamectin, cepharantine, mefloquine hydrochloride, nitazoxanide and ivermectin have in-vitro antiviral activity against SARS-CoV-2. Both the anti-malarial drugs, chloroquine and hydroxychloroquine are immunomodulators and down regulate cytokine production which mitigate the effect of SARS-CoV-2 in the target organs such as lungs, heart, liver and gut. However, the results of preliminary large scale randomized controlled trials have failed to show any survival benefit of these two drugs in COVID-19 [50]. The triple combination of cepharantine, selamectin and mefloquine hydrochloride has recently been shown to inhibit the infection of simian vero E6 cells with pangolin coronavirus GC_ P2V/2017/Guangxi (GX_P2V), where S protein shares 92.2% amino acid identity with that of SARS-CoV-2 and thus it is identified as a candidate drug combination against SARS-CoV-2 infection [4, 51]. The antiparasitic drug nitazoxanide (NTZ) which has been found to have antiviral activity against different viral infections, especially coronaviruses exhibited in-vitro inhibition of SARS-CoV-2 at a small micromolar concentration. It also suppresses the production of cytokines emphasizing its potential to manage COVID-19 induced cytokine storm, adding to its high safety records for toxicity [52]. The FDA approved anti-parasitic drug ivermectin which is used to treat various parasitic infections such as river blindness, head lice, scabies, lymphatic filariasis, ascariasis, entrobiasis, strongyloidiasis and trichuriasis has been found to have anti-cancer, anti-diabetic and anti-viral properties besides their antihelminthic and insecticidal properties [2,53]. Ivermectin’s broad spectrum antiviral activity in-vitro is effective against both DNA and RNA viruses and its mechanism of action lies in inhibiting the nuclear import of viral proteins [2,54,55]. Several SARS-CoV-2 related proteins are regulated by ivermectin and thus offers more potentiality to improve global public health, and it can effectively inhibit the replication of SARSCoV- 2 in-vitro [56, 57]. Ivermectin with a single addition to VerohSLAM cells post infection with SARS-CoV-2 was able to affect 5000 fold reduction in viral RNA at 48 hours [54]. Drug combination of ivermectin and hydroxychloroquine could be a suitable combination therapy for the prophylaxis or treatment of COVID-19. This combination therapy might show a consequential and synergistic effect with both viral entry and viral replication for COVID-19 [55, 56].
A further detailed elucidation on the molecular properties and the interaction profiles of the top 10 antiparasitic drug inhibitors will owe to a better understanding on their antiviral action against SARSCoV- 2.
Selamectin
Selamectin (CID9578507) is a topical parasiticide and antihelminthic drug used to treat and prevent infections of heartworms, fleas, ear mites, sarcoptic mange, hookworms and roundworms [23]. The molecule having a molecular weight of 769.96 g/mol, 12 hydrogen bond acceptors (HA) and 3 hydrogen bond donors (HD) forms 2 hydrogen bonds, 2 electrostatic and 3 hydrophobic interactions with the residues PHE 464, and GLU465 in the site-1 of the spike(S) protein’s receptor binding domain (RBD) with a binding affinity of -11.2 kcal/mol. Figure 4 represent the docked pose of selamectin with the target protein in 3D and in 2D depiction (Figure 5a and 5b).

Figure 5: Interaction profile of selamectin with RBD. (A) 3D visualization of the interactions (B) 2D depiction of the interactions.
Ivermectin
Ivermectin (CID6321424) is an orally bioavailable macrocyclic lactone derived from Streptomyces avermitilis with potential antiparasitic activities against several parasitic nematodes, scabies and is extensively used in the treatment of onchocerciasis (liver blindness) [23]. Its formation of 3 hydrogen bonds, 3 electrostatic and 3 hydrophilic interactions with residues PRO337, GLU340, ARG355, ARG357, TYR396 and ARG466 in the site-2 of RBD and with a binding affinity of -9.3 kcal/mol is attributed to its possession of 14 HA and 3HD in its entire structure with a molecular weight of 857.09 g/mol (Figure 6a and 6b).

Figure 6: Interaction profile of ivermectin with RBD. (A) 3D visualization of the interactions. (B) 2D depiction of the interactions.
Artefenomel
Artefenomel(CID24999143) which has been inspected for the treatment of malaria [23], has a molecular weight of 469.61g/mol with 6 HA. It forms 3 hydrogen bonds, 1 electrostatic and 4 hydrophobic interaction with residues TYR396, PHE429, PHE464, GLU465 and ARG466 on the site-3 of RBD with a binding affinity of -8.5 kcal/mol (Figure7a and7b).

Figure 7: Interaction profile of artefenomel with RBD. (A) 3D visualization of the interactions. (B) 2D depiction of the interactions.
Moxidectin
Moxidectin (CID9832912) which is classified as a second generation macrocyclic lactone is derived from Streptomyces cyanogriseus with potential antiparasitic activity. The drug being a potent broad spectrum endectocide (anti-parasitic that is active against both endo and ecto parasites) against nematodes, insects and ascari was first used in cattle followed by an approved use in general animals. It is a semisynthetic methoxine derivative of nemadectin which is a 16 member pentacyclic lactone of the milbemycin class [23]. The molecule forms 2 hydrophobic interactions with residues LYS417 and LEU455 in the site-2 with a binding affinity of -8.5 kcal/ mol. The molecule with a total weight of 639.82 g/mol has 9 HA and 2 HD in its structure (Figure 8a and 8b).

Figure 8: Interaction profile of moxidectin with RBD. (A) 3D visualization of the interactions. (B) 2D depiction of the interactions.
Posaconazole
Posaconazole (CID468595) acting as a trypanocidal drug is a broad spectrum, second generation, triazole compound with antifungal activity against aspergillosis and candida. Compared to other azole antifungals, posaconazole is a significantly more potent inhibitor of sterol 14-alpha demethylase which is a cytochrome P450 dependent enzyme [23]. It has a molecular weight of 700.78 g/mol with 9 HA and 1 HD forming a hydrogen bond, 2 electrostatic and 2 hydrophobic interactions with residues TYR421, PHE456 and TYR489 on the site-2 of RBD with a binding affinity of -8.3 kcal/mol (Figure 9).

Figure 9: Interaction profile of posaconazole with RBD- 3D visualization of the interactions. The 2D depiction of the interactions of posaconazole is not available since the ligand is not a single fragment.
Imidocarb
Imidocarb (CID21389) is an antiprotozoal drug which is a urea derivative used in veterinary medicine for the treatment of infection with Babesia and other parasites [23]. It forms 6 hydrogen bonds and 3 hydrophobic interactions with residues LYS458, GLU471, ILE472, TYR473, GLN474, ALA475, THR478 and TYR489 on the site-3 of RBD with a binding affinity of -8 kcal/mol. This large number of hydrogen bond formation is due to the presence of 3 HA and 4 HD in its structure with a total molecular weight of 348.4 kcal/mol (Figure 10a and 10b).

Figure 10: Interaction profile of imidocarb with RBD. (A) 3D visualization of the interactions. (B) 2D depiction of the interactions.
Piperaquine
Piperaquine(CID122262) acting as an antimalarial agent is an aminoquinolone that is 1,3-di(piperazin-1-yl)propane in which the nitrogen of position 4 of each of piperazine moieties is replaced by a 7-chloro quinolon-4-yl group [23]. The drug with a molecular weight of 535.51 g/mol and 4 HA in its structure forms 4 hydrogen bonds and 7 hydrophobic interactions with the residues ILE418, TYR421, PHE456, ARG457, LYS458 and ALA475 on the site-3 of RBD with a binding affinity of -7.9 kcal/mol (Figure 11a and 11b).

Figure 11: Interaction profile of piperaquine with RBD. (A) 3D visualization of the interactions. (B) 2D depiction of the interactions.
Cepharantine
Cepharantine(CID7098680) is an anti-inflammatory alkaloid derived from Stephania cepharantha hayata [4]. The molecule with a total molecular weight of 606.71 g/mol having 8 HA forms 2 hydrogen bonds, 2 electrostatic and 3 hydrophobic interactions with the residues ARG355, TYR396, ARG429, GLU465 and PHE515 on the RBD’s site-2 with a binding affinity of -7,9 kcal/mol (Figure 12a and 12b).

Figure 12: Interaction profile of cepharantine with RBD. (A) 3D visualization of the interactions. (B) 2D depiction of the interactions.
Betulinic acid
Betulinic acid(CID64971) exhibits anti-HIV, anti-malarial, antineoplastic and anti-inflammatory properties and is a pentacyclic lupane-type triterpene derivative of betulin, isolated from the bark of Betula alba [23]. It forms a hydrophobic interaction with the residue PHE429 on the site-2 with a binding affinity of -7.9 kcal/mol. It has a total molecular weight of 456.7 kcal/mol with 3 HA and 2HD (Figure 13a and 13b).

Figure 13: Interaction profile of betulinic acid with RBD. (A) 3D visualization of the interactions. (B) 2D depiction of the interactions.
Atovaquone
Atovaquone (CID74989) is a synthetic hydroxy naphthoquinone with antiprotozoal activity used for the prevention and treatment of Pneumocystis jevorici (formerly carinii) pneumonia. It is also used in combination with proguanil for the prevention and treatment of Plasmodium falciparum, thus acting as an antimalarial agent [23]. The compound with a total molecular weight of 366.84 g/mol, 3 HA and 1 HD in its structure forms a hydrogen bond and 4 hydrophobic interactions with residues TYR396, PHE464, SER514 and GLU516 on the site-1 of RBD with a binding affinity of -7.7 kcal/mol (Figure 14a and 14b).

Figure 14: Interaction profile of atovaquone with RBD. (A) 3D visualization of the interactions. (B) 2D depiction of the interactions.
Conclusions
It is well known that in order to bring the SARS-CoV-2 infection under control; a drug would just be a temporary remedy and a suitable vaccine could only become a radically permanent solution. Though the repurposed drugs are short-term reliefs and cannot become permanent solutions to the pandemic, the advantage of getting easy and immediate access to the drugs, following their high safety records in terms of toxicity, outweighs the demerit of the drug’s sustenance.
This COVID-19 pandemic disconcerted the world countries with millions of deaths and an economic crisis that took place as never before. And most importantly, rumours and unnecessary chaos in social media were spreading in exponential rates than did the virus in its lytic cycle of multiplication. The coronavirus kindled the roots for fear and panic in the minds of every individual including the scientific professionals and medical practitioners. And it’s quite fascinating to conceptualize the world’s after-math of this COVID-19 pandemic. We are now combating against this corona virus with a serious notion and for sure we will set foot on the pinnacle though it may not occur in the nearest future. But then, our livelihoods after that won’t be the same as it was before and we will be colonizing an entirely new conflicting world for survival. The virus is global and hence eventually the reverberation of the outbreak is also global. We may be deprived of some stereotyped practices, while we may experiment and stick to the guns of novel techniques and courses of action, yearning for a reformed livelihood. For sure, life wouldn’t be a child’s play to confront with in the wake of this catastrophe. We would have to square up to the hassles and toils following this prolonged period of curfew and quarantine. And we would have to wrestle and come to blows with the suddenly mutated environment. But all these things could become fortunately possible, only when we, the Homo sapiens as the higher animals on earth, become flexible with the changes that occur in the rediscovered world. Now after very long months of quarantine, face masks, sanitizers and online lifestyle, everything is so weird seemingly on taking a look aback. But it’s time for us to realize that this is just the beginning of a new normal epoch for years to come and there will survive the fittest of the races who find their quickest adaptation towards this. We humans are resilient and we can learn to thrive in our new normal if we have the mindset and the resources we need to adapt. Thus with confident and hopeful insights, let’s stand up to the new normal world and we are anticipating a quick cure for the pandemic infection in the form of a repurposed or virus-specific drug that would soon happen by the efforts of the medical and scientific community!
Acknowledgment
The authors thank staff within the Microbiology Laboratory for their kind helps. The current study was financially supported by a grant from the Deputy Dean for Research, Tehran University of Medical Sciences (grant no. 97-02-27- 39571).
Conflict of Interest
The authors declare no conflict of interest.
References
- UNNI S, Aouti S, Balasundaram P (2020) Identification of a Potent Inhibitor Targeting the Spike Protein of Pandemic Human Coronavirus. SARS-CoV-2 by Computational Methods, 45(1): 130.
- Dixit A, Yadav R, Singh AV (2020) Ivermectin: Potential role as repurposed drug for covid-19. Malaysian J Med Sci, 27(4): 154–158.
- Tahir ul Qamar M, Alqahtani SM, Alamri MA, Chen LL (2020) Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal, 10 (4): 313–319.
- Mckee DL, Sternberg A, Stange U, Laufer S, Naujokat C, et al. (2020) Since January 2020 Elsevier has created a COVID-19 resource centre with free information in English and Mandarin on the novel coronavirus COVID- 19 . The COVID-19 resource centre is hosted on Elsevier Connect , the company ’ s public news and information.
- Tiwari V, Beer JC, Sankaranarayanan NV, Swanson-mungerson M, Desai UR, et al. (2020) Discovering small-molecule therapeutics against SARS- CoV-2, 25(8): 1535-1544.
- Adedeji AO, Severson W, Jonsson C, Singh K, Weiss SR, et al. (2013) Novel Inhibitors of Severe Acute Respiratory Syndrome Coronavirus Entry That Act by Three Distinct Mechanisms. J Virol, 87(4): 8017–8028.
- Xia S, Liu M, Wang C, Xu W, Lan Q, et al. (2020) Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res, 30(4): 343–355.
- Pink R, Hudson A, Mouriès MA, Bendig M (2005) Opportunities and challenges in antiparasitic drug discovery. Nat Re Drug Discov, 4: 727–740.
- Sethi A, Joshi K, Sasikala K, Alvala M, (2020) Molecular Docking in Modern Drug Discovery: Principles and Recent Applications. Drug Discov Dev New Adv, 1–21.
- Yang H, Sun L, Li W, Liu G, Tang Y, et al. (2018) In Silico Prediction of Chemical Toxicity for Drug Design Using Machine Learning Methods and Structural Alerts Front Chem, 6: 1–12.
- Gaillard T (2018) Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J Chem Inf Model, 58 (8): 1697–1706.
- Jaghoori MM, Bleijlevens B, Olabarriaga SD (2016) 1001 Ways to run AutoDock Vina for virtual screening. J Comput Aided Mol Des, 30 (3): 237– 249.
- Vieira TF, Sousa SF (2019) Comparing AutoDock and Vina in ligand/decoy discrimination for virtual screening. Appl Sci, 9(21):4538.
- Nguyen NT, Nguyen TH, Pham TNH, Huy NT, Van Bay M, et al. (2020) Autodock Vina Adopts More Accurate Binding Poses but Autodock4 Forms Better Binding Affinity. J Chem Inf Model, 60(1): 204–211.
- Basu A, Sarkar A, Maulik U,(2020) Molecular docking study of potential phytochemicals and their effects on the complex of SARS-CoV2 spike protein and human ACE2. Sci Rep, 10: 1–15.
- Saxena A (2020) Drug targets for COVID-19 therapeutics: Ongoing global efforts. J Biosci. 45(1): 87.
- Pagadala NS, Syed K, Tuszynski J (2017) Software for molecular docking: a review. Biophys Rev, 9 (2): 91–102.
- Heimfarth L, Serafini MR, Martins-Filho PR, Quintans J. de SS, Quintans- Júnior LJ, et al. (2020) Drug repurposing and cytokine management in response to COVID-19: A review. Int Immunopharmacol, 88: 106947.
- Papich MG (2016) Doramectin. Saunders Handb Vet Drugs, 266–268.
- Trott O, Olson AJ (2009) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem.
- Prieto-Martínez FD, Arciniega M, Medina-Franco JL (2018) Acoplamiento Molecular: Avances Recientes y Retos. TIP Rev Espec En Ciencias Químico- Biológicas, 21:0–23.
- Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, et al. (2018) J DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res, 46(D1): 074–D1082.
- Kim S, Chen J, Cheng T, Gindulyte A, He J, et al. (2019) PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res, 47: D1102– D1109.
- Boyle NMO, Banck M, James CA, Morley C, Vandermeersch T, et al. (2011) Open Babel: An open chemical toolbox - 1758-2946-3-33, 1–14.
- Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, et al. (2015) GenBank. Nucleic Acids Res, 43(D1): D30–D35.
- Berman H, Henrick K, Nakamura H (2003) Announcing the worldwide Protein Data Bank. Nat Struct Biol, 10(12): 980.
- Yang J, Yan R, Roy A, Xu D, Poisson J, et al. (2014) The I-TASSER suite: Protein structure and function prediction. Nat Methods, 12(1): 7–8.
- Zhang C, Zheng W, Huang X, Bell EW, Zhou X, et al. (2020) Protein Structure and Sequence Reanalysis of 2019-nCoV Genome Refutes Snakes as Its Intermediate Host and the Unique Similarity between Its Spike Protein Insertions and HIV-1. J Proteome Res, 19(4):1351–1360.
- Yuan S, Chan HCS, Hu Z (2017) Using PyMOL as a platform for computational drug design, Wiley Interdiscip. Rev Comput Mol Sci, 7: 1–10.
- Roy A, Kucukural A, Zhang Y (2010) I-TASSER: A unified platform for automated protein structure and function prediction. Nat Protoc, 5(4): 725– 738.
- Wu S, Skolnick J, Zhang Y (2007) Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol, 5: 1–10.
- Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics, 9: 1–8.
- Yang J, Zhang Y (2015) I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res, 43(W1): W174–W181.
- Zheng W, Li Y, Zhang C, Pearce R, Mortuza SM, et al. (2019) Deep-learning contact-map guided protein structure prediction in CASP13. Proteins Struct Funct Bioinforma, 87(12): 1149–1164.
- Li Y, Zhang C, Bell EW, Yu DJ, Zhang Y, et al. (2019) Ensembling multiple raw coevolutionary features with deep residual neural networks for contact-map prediction in CASP13. Proteins Struct. Funct Bioinforma, 87(12): 1082–1091.
- Zhang Y, Skolnick J (2004) Scoring function for automated assessment of protein structure template quality. Proteins Struct Funct Genet, 57(4): 702–710.
- Xu J, Zhang Y (2010) How significant is a protein structure similarity with TM-score = 0.5?. Bioinformatics, 26(7): 889–895.
- Li Y, Hu J, Zhang C, Yu DJ (2019) ResPRE: High-accuracy protein contact prediction by coupling precision matrix with deep residual neural networks. Bioinformatics, 35(22): 4647–4655.
- Yang J, Zhang Y (2015) Protein Structure and Function Prediction Using I-TASSER. Curr Protoc Bioinforma, 52: 5.8.1-5.8.15.
- Goodsell DS, Olson AJ (1990) Automated docking of substrates to proteins by simulated annealing, Proteins Struct. Funct Bioinforma, 8(3): 195–202.
- Ton AT, Gentile F, Hsing M, Ban F, Cherkasov A, et al. (2020) Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol Inform, 39(8): 1–8.
- Baig AM, Khaleeq A, Syeda H (2020) Docking Prediction of Amantadine in the Receptor Binding Domain of Spike Protein of SARS-CoV-2, ACS Pharmacol. Transl Sci, 3(6): 1430-1433.
- Pandey P, Rane JS, Chatterjee A, Kumar A, Khan R, et al. (2020) Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: an in silico study for drug development. J Biomol Struct Dyn, 1–11.
- Ibrahim IM, Abdelmalek DH, Elshahat ME, Elfiky AA (2020) prediction. 80(5): 554-562.
- Liu S, Xiao G, Chen Y, He Y, Niu J, et al. (2004) Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: Implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet, 363(94114): 938–947.
- Goswami T, Bagchi B (2020) Molecular Docking study of Receptor Binding Domain of SARS-CoV-2 Spike Glycoprotein with Saikosaponin. a Triterpenoid Natural Product.
- Othman H, Bouslama Z, Brandenburg JT, da Rocha J, Hamdi Y, et al.(2020) Interaction of the spike protein RBD from SARS-CoV-2 with ACE2: Similarity with SARS-CoV, hot-spot analysis and effect of the receptor polymorphism. Biochem Biophys Res Commun, 527(3): 702–708.
- Drożdżal S, Rosik J, Lechowicz K, Machaj F, Kotfis K, et al. (2020) FDA approved drugs with pharmacotherapeutic potential for SARS-CoV-2 (COVID-19) therapy. Drug Resist Updat, 53: 100719.
- Chary MA, Barbuto AF, Izadmehr S, Hayes BD, Burns MM, et al. (2020) COVID-19: Therapeutics and Their Toxicities. J Med Toxicol, 16(3): 284–294.
- Khuroo MS (2020) Chloroquine and hydroxychloroquine in coronavirus disease 2019 (COVID-19). Facts, fiction and the hype: a critical appraisal. Int J Antimicrob Agents, 56(3): 106101.
- Fan HH, Wang LQ, Liu WL, An XP, Liu ZD, et al. (2020) Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus-related coronavirus model. Chin Med J,133(9): 1051–1056.
- Mahmoud DB, Shitu Z, Mostafa A (2020) Drug repurposing of nitazoxanide: can it be an effective therapy for COVID-19?. J Genet Eng Biotechnol, 18(1): 35.
- Batiha GES, Alqahtani A, Ilesanmi OB, Saati AA, El-Mleeh A, et al. (2020) A vermectin derivatives, pharmacokinetics, therapeutic and toxic dosages, mechanism of action, and their biological effects. Pharmaceuticals, 13(8): 196.
- Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM, et al. (2020) The FDA- approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res, 178:104787.
- Mudatsir M, Yufika A, Nainu F, Frediansyah A, Megawati D, et al. (2020) Antiviral activity of ivermectin against SARS-CoV-2: An old-fashioned dog with a new trick‒A literature review. Sci Pharm, 88(3): 1–8.
- Li N, Zhao L, Zhan X (2020) Quantitative proteomics reveals a broad- spectrum antiviral property of ivermectin, benefiting for COVID-19 treatment. J Cell Physiol, 236(4): 2959-2975.
- Peña-Silva R, Duffull SB, Steer AC, Jaramillo-Rincon SX, Gwee A, et al. (2020) Pharmacokinetic considerations on the repurposing of ivermectin for treatment of COVID-19. Br J Clin Pharmacol, 87(3): 1589-1590.