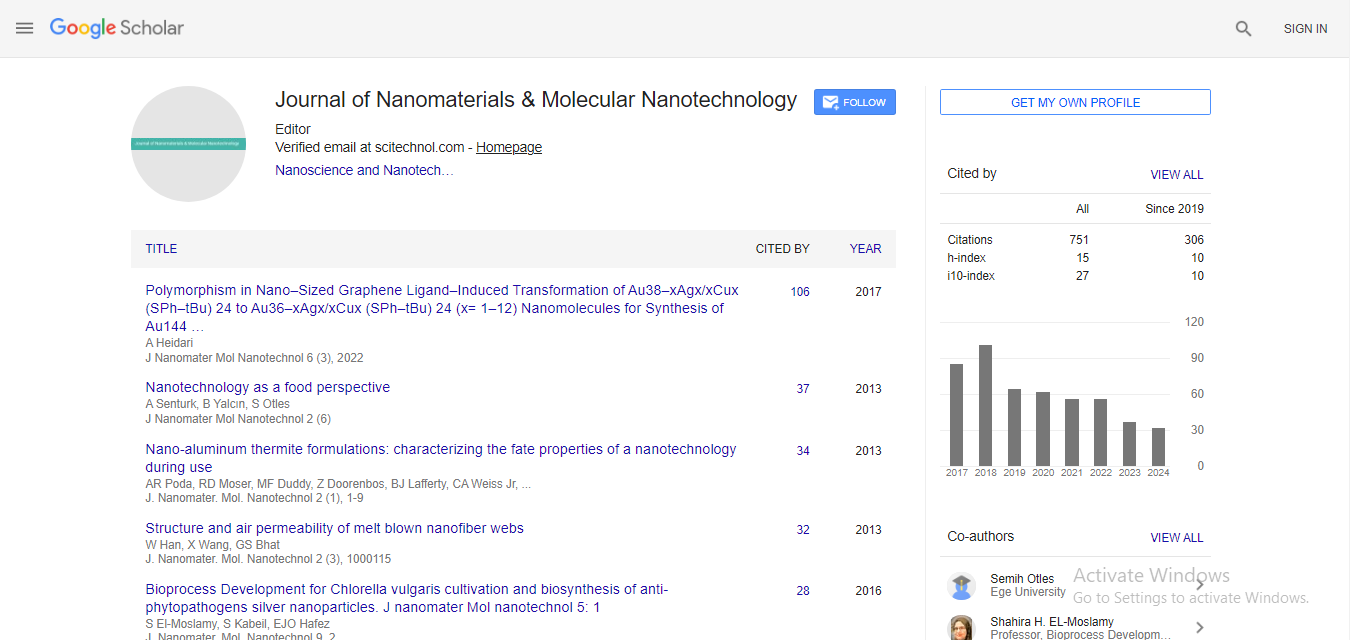
Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Nanomater Mol Nanotechnol S Vol: 7 Issue: 5
Nanoparticle Augmented Radiotherapy using Titanium Oxide Nanoparticles
Gareth Wakefield*, Martin Gardener, Matt Stock and Megan Adair
Xerion Healthcare Ltd, Cherwell Innovation Centre, 77 Heyford Park, Upper Heyford, Oxfordshire OX25 5HD, UK
*Corresponding Author : Gareth Wakefield
Xerion Healthcare Ltd, Cherwell Innovation Centre, 77 Heyford Park, Upper Heyford, Oxfordshire OX25 5HD, UK
Tel: +44 01869 238305
E-mail: gareth.wakefield@xerionhealthcare.co.uk
Received: June 15, 2018 Accepted: July 19, 2018 Published: July 25, 2018
Citation: Wakefield G, Gardener M, Stock M, Adair M (2018) Nanoparticle Augmented Radiotherapy using Titanium Oxide Nanoparticles. J Nanomater Mol Nanotechnol S6: 002. doi: 10.4172/2324-8777.S6-002
Abstract
Titanium oxide is a photoactive material that generates hydroxyl free radicals via water splitting. When doped with rare earth ions titanium oxide nanoparticles are activated by X-rays and X-ray generated electrons and are used to enhance radiotherapy treatment of solid tumours. As the nanoparticles generate free radicals by water splitting the presence of molecular oxygen is not required and aggressive hypoxic tumours may be targeted. A clonogenic assay of radio resistant pancreatic cancer (PANC- 1) cells shows a radiotherapy dose enhancement factor of 1.9 at clinically relevant nanoparticle loadings. A fast growing oropharyngeal cancer (FaDu) xenograft demonstrates that rare earth doped titanium oxide nanoparticles delivered by intratumoural injection disperse throughout the tumour, being taken up by cancer cells and undergoing passive accumulation in the Golgi apparatus. Incident radiotherapy activates the nanoparticles to produce hydroxyl free radicals, destroying the Golgi apparatus, and inducing tumour cell apoptosis. This results in a reduction in proliferating cancer cells and a consequent reduction in tumour regrowth rate by a factor of 3.8. There is no increase in systemic toxicity when using nanoparticles in addition to radiotherapy. Rare earth doped titanium oxide nanoparticles therefore represent a novel approach to tumour treatment via destruction of the cells Golgi apparatus during radiotherapy.
Keywords: Titanium oxide; Cancer; Xenograft; Golgi apparatus; Radiotherapy; Rare earths
Abbreviations
BrdU: Bromodeoxyuridine or 5-bromo-2’-deoxyuridine; TEM: Transmission Electron Microscopy; DEF: Dose Enhancement Factor; FaDu: Human derived oropharyngeal carcinoma immortal cell line; PANC-1: Human derived pancreatic adenocarcinoma immortal cell line
Introductions
Cancer represents the second most important cause of death and morbidity in the EU, with an estimated 3.5 m cases and 1.9 m deaths per annum. The proportion of cancer patients in which external beam radiotherapy is indicated is 52% however actual utilisation rates suggest a shortfall in radiotherapy provision [1]. For example, in England an increase in radiotherapy provision of 50% would be required to match demand for the treatment, equivalent to 36,000 cases per annum. Demand is primarily driven by breast (28% of attendances), head and neck (9%), lung (13%) and prostate (29%), which together account for 79% of radiotherapy treatments. Improved radiotherapy outcomes, including dose de-escalation, dose optimisation and reduction of localised reoccurrence in these indications would have the greatest effect on overall radiotherapy demand and utilisation [2]. Radiotherapy is second only to surgery in its cure rates and is involved in 40% of cases where patients are cured of their cancer. It is the primary treatment modality used in 16% of patients, often in combination with chemotherapy or immunotherapy [3]. By comparison, chemotherapy is the principal modality in only 2% of curative treatments with little progress having been made over the past few decades. An overall survival benefit of less than 5% has been achieved by adjuvant chemotherapy treatment of breast, colon and head and neck cancers whereas lung cancer mean survival is only increased by 2 months following treatment with Bevacizumab (Avastin) [4]. The EU estimates that radiotherapy costs €3000 per patient compared with €7000 for surgery and, on average, €17000 for chemotherapy [5]. This is highly dependent on indication and whether treatment intent is curative or palliative. However, even for the highest curative doses (60-70 Gy) employed in the treatment of head and neck cancer, the costs only amount to between €8250 and €9625 per patient [6].
Radiotherapy works by targeting a beam of high energy photons, typically in the MeV energy range, at the tumour site. Photons may interact with the tumour cell either directly, by DNA absorption, or indirectly as the X-ray scatters off water molecules in the tumour. Indirect scattering results in >90% of the incident energy being transferred to an electron. This electron scatters off other nearby electrons causing a cascade effect where a field of progressively less energetic electrons are generated, finally resulting in interaction with molecular oxygen and consequent generation of superoxide free radicals. Superoxide free radicals then induce cancer cell damage and apoptosis [7]. Of course, damage to normal cells will also occur as a result of the X-ray and consequently the radiation beam is carefully targeted and shaped to get as much of the energy into the tumour as possible. Whilst normal cells can repair themselves more efficiently against free radical damage than cancer cells there is not much difference in the ability of cancerous and normal tissue to absorb X-rays. Consequently, X-ray dose is determined by the tolerance of the surrounding normal tissues rather than the dose required controlling tumour growth. In principle it is possible to treat any tumour with radiotherapy but in practice this would mean damaging the patient. Too little radiation will have little effect whereas too much can lead to significant side effects such as permanent damage to vital organs or radiation burns. There is therefore what is known as a ‘therapeutic window’ for radiotherapy treatment which is constantly being extended by the implementation of more complex radiotherapy delivery systems. However the current arc of improvements are being fuelled by advances in radiation physics, instrumentation and computing power and are starting to reach technological limits. Complicating this is the fact that various tumour types respond differently to radiotherapy, being more or less radiosensitive or radio resistant, as do various other organs in the body [8].
Solid tumours are highly heterogeneous with vascularised, normally oxygenated regions, oxygen deficient hypoxic regions and necrotic cores. The hypoxic region is extremely important in the treatment of cancer. In order to support continuing rapid cell proliferation oxygen deficient cancer cells alter their metabolism [9]. The hypoxic region generally evades traditional chemotherapy compounds and is also much less responsive than normally oxygenated, or normoxic, cells to the effects of radiotherapy since molecular oxygen is required to form superoxide free radicals. Radiotherapy is 2-3 times less effective in hypoxic tumour regions. Fractionated dosing is commonly used to at least partially address this issue. Following the sudden death of a proportion of well oxygenated cells, surviving hypoxic cells re-oxygenate and become susceptible to further radiotherapy fractions. These cells also have enhanced radio resistance which compounds the problem. This has significant clinical repercussions since hypoxia is associated with the most aggressive tumour phenotypes [10].
One way of enhancing free radical generation during radiotherapy is to use nanoparticles introduced into the tumour to interact with incident X-ray photons and photo generated electrons. To date nanoparticles located within the tumour in this way have been designed to augment the effects of radiotherapy by enhancing the scattering of X-ray generated photoelectrons, effectively slowing them down to get to the final superoxide generating event with a shorter path length. This has the overall effect of making the free radical generation volume from any principal X-ray scattering event within the tumour smaller. As the overall energy dissipated is equivalent the free radical concentration within this volume is increased. As the interaction cross-section is dependent on atomic number (Z) most work in this area has concentrated on high-Z gold nanoparticles. A number of studies have shown that gold nanoparticles are effective at tumour control and show comparable sensitivity at both kilovoltage and megavoltage X-ray energy [11,12]. More recently, hafnium oxide nanoparticles have been employed in a similar fashion exhibiting radiotherapy dose enhancement against a variety of radiosensitive and radio resistant cancer cell lines in clonogenic assays [13].
Titanium oxide is a well-known, non-toxic, photoactive material that generates hydroxyl free radicals by water splitting under ultraviolet light [14]. This property is related to the unusual electronic band structure of the anatase phase of titanium oxide in which the direct and indirect transition energies are essentially degenerate with a small energy barrier between them. As such, following UV excitation, there exist a significant population of conduction band electrons occupying the relatively long lifetime indirect transition, with a corresponding hole at the top of the valence band. In titanium oxide nanoparticles the charged particles wave functions can occupy a relatively high proportion of the particle volume and de-excitation via localised surface states is energetically favourable. Consequently, the conduction band electrons, eCb-, and valence band holes, hVb+, can de-excite creating surface free radicals following the equations:

Titanium oxide can generate hydroxyl or superoxide radicals depending on whether the electron or hole is used, but a critical aspect for tumour treatment is that the hole mediated reaction does not require oxygen and will be just as effective in a hypoxic tumour region as under normoxic conditions [15,16]. Doping rare earth ions into titanium oxide nanoparticles results in strong interactions with X-ray generated electrons due to the high atomic number of rare earth ions and the consequent free radical production induces cell apoptosis under external beam radiotherapy. In this report we demonstrate that the use of rare earth doped titanium oxide nanoparticles is effective at enhancing radiotherapy efficacy in radio resistant cell lines and murine xenograft models. Nanoparticles of titanium oxide doped with rare earth ions act in a novel way to current high atomic mass nanoparticle technologies radiotherapy and have been shown to be effective in vitro and in vivo in previous reports [17]. This report extends this work to elucidate the mechanism of cancer cell death, provide dose enhancement factors for pancreatic PANC-1 cell lines and model regrowth rates from oropharyngeal FaDu xenograft models.
Materials and Methods
Rare earth doped titanium oxide nanoparticles
Nanoparticles of rare earth doped titanium oxide were used as supplied by Xerion Healthcare, Cherwell Innovation Centre, 77 Heyford Park, Upper Heyford, OX25 5HD, UK. The particles have a mean size (d50) of 50 nm (d90=100) to allow effective transport through the tumour and uptake via endocytosis into cancer cells [18] and are doped with up to 9 wt% rare earth ions.
In vitro (PANC-1) clonogenic assay
A clonogenic assay is an in vitro cell survival assay based on the ability of a single cell to grow into a colony and is the in vitro method of choice to determine cell death after treatment with ionising radiation. The assay follows a standard protocol [19]. Briefly, a radio resistant pancreatic immortal cell line (PANC-1), derived from a pancreatic ductal adenocarcinoma in a 56-year-old Caucasian male, was chosen as the test cell line [20]. PANC-1 cells were thawed, cultured and seeded at 2000 cells per well in triplicate. Nanoparticles were added at 6.25 mg (57 μM) to each well and incubated for 24 h prior to irradiation. Cells were irradiated in the presence of control vehicle or the nanoparticle dispersion at 0, 1, 2, 3, 4, 5 and 6 Grays using an XStrahl RS320 cabinet irradiator operated at 300 KV, 10 mA delivering at a dose rate of 0.818 Gy.min-1. The plates were incubated at 37°C, 5% CO2 and 95% RH and harvested on day 6. Upon harvesting cells were stained with crystal violet, washed and air dried. Colonies were scored and survival fractions were determined in comparison to 0 Gy groups. Dose enhancement factors were calculated at 10% survival fraction. The dose enhancement factor is defined as:

Dose enhancement factors were also calculated as a function of dose dependence by calculating the ratio of the two survival fractions at each applied radiotherapy dose.
In vivo (FaDu) xenograft assay
The ectopic tumour xenograft model, in which immortal cell lines derived from human tumours are subcutaneously implanted in the flank of immunocompromised mice, is the standard model of cancer used for validation and assessment purposes [21]. FaDu cancer cells, derived from an HPV negative squamous cell carcinoma of the oropharynx, were used as a radio resistant xenograft model cancer [22]. Female CD-1 nude mice were purchased at 4-6 weeks of age and, following 1 week stabilisation, xenografted with 1 × 106 FaDu cells on one flank. Tumours were left to grow until the mean tumour volume, as measured with callipers, reached 100-150 mm3. Mice were randomised into four groups of n=12 animals. Radiation was delivered using an Xstrahl RS320 cabinet irradiator operating at 300 KV, 10 mA delivering at a dose rate of 0.696 Gy.min-1. The radiation tube has additional filtration to give a radiation quality of 2.3 mm Cu half-value layer. Mice were placed in a Perspex jig positioned at a distance of 700 mm from the focus of the X-ray tube. The body was lead shielded except for a 1 cm hole above the tumour site before radiation is delivered as a single dose.
Group 1 were irradiated for 5 days at 2 Gy over 2 cycles (days 1-5 and 8-12) to a total of 20 Gy.
Group 2 were injected intratumourally with 20 μl of 6.25 mg.ml-1 rare earth doped titanium oxide nanoparticles dispersed in phosphate buffered saline on Days 1 and 8 and were irradiated for 5 days at 2 Gy over 2 cycles (days 1-5 and 8-12) to a total of 20 Gy.
Group 3 underwent sham irradiation over two cycles on Days 1-5 and 8-12.
Group 4 were injected intratumourally with 20 μl of 6.25 mg.ml-1 rare earth doped titanium oxide nanoparticles dispersed in phosphate buffered saline on Days 1 and 8 and underwent sham irradiation over two cycles on Days 1-5 and 8-12.
All animals were anaesthetised during the irradiation process (including sham irradiation) using injectable anaesthetics (ketamine and xylazine). Tumours and animal weight were measured 3 times weekly. Animal weight was used as a proxy for systemic toxicity of the nanoparticles and radiotherapy treatment. Two animals from each group were culled two days post final treatment. The remaining animals were allowed to grow out for tumour volume analysis until day 35 of the study or until the tumours reach 1 cm3. Forty minutes prior to sacrifice mice were injected with 0.5 ml of a 20 mg.ml-1 solution of bromodeoxyuridine (BrdU). BrdU is a synthetic nucleoside which is an analogue of thymidine and is incorporated into the DNA of proliferating cells during the S-phase, substituting for thymidine. Following removal, tumours were split in half; one half being fixed in formalin and processed to provide paraffin embedded tissue block for histology while the second half was fixed in glutaraldehyde and dehydrated prior to microtoming for TEM analysis. Following processing into a paraffin block, samples for BrdU histology were dewaxed in xylene. Endogenous peroxidase was blocked with 1% H2O2 prior to washing and incubation with rabbit serum to block non-specific background. An antibody specific for BrdU, rat anti-BrdU (AbD Serotec OBT0030S; clone BU1/75 (ICR1)) was applied and incubated for 1 h. A secondary antibody, peroxidase-conjugated rabbit anti-rat IgG (Dako P0450) at 1% concentration in 5% mouse serum, was then incubated for 1 h. After washing, labelling was visualised with DAB (3, 3 -diaminobenzidine) (Vector Impact SK-4105) for 5 minutes before being counterstained with haematoxylin and permanently mounted. All in vitro and in vivo work was undertaken by Epistem Ltd, Manchester, UK. Samples for TEM were analysed at the cryo-TEM JEOL2200-FS facility at the University of Warwick.
Results and Discussion
PANC-1 In vitro clonogenic assay
Clonogenic studies were undertaken at a dose of 6.25 mg (57 μM) nanoparticles per well, equivalent to the concentration of nanoparticles used in in vivo studies and what would be considered to be clinically relevant volumes and concentrations for intratumoural injection (i.e. <5% total tumour volume and <5 wt% nanoparticle concentration). PANC-1 cells show strong dose dependent radiotherapy enhancement as shown in Figure 1. Figure 1A gives cell survival curves of PANC-1 cells with and without nanoparticles as a function of radiotherapy dose; at 90% cell death the dose enhancement factor (DEF) is 1.9. The cell death ratio as a function of dose is given in Figure 1B. Dose enhancement factors range from 2.2 at 1 Gy to 8.6 at 4 Gy.

Figure 1: A) PANC-1 pancreatic cancer cell survival curves. Clonogenic assays were incubated with 57 µM rare earth doped titanium oxide nanoparticles for 24 h prior to irradiation and surviving colonies counted. The Dose Enhancement Factor (DEF) at 90% cell death is 1.9, B) PANC-1 DEF as a function of increasing dose (R2=0.98).
Previous work describing clonogenic assays of radio sensitizers for PANC-1 cells has shown mixed results. Histamine, which is effective as a radio sensitizer for breast cancer cells shows no significant radio sensitization effect on the PANC-1 cell line [23]. In contrast, metformin in PANC-1 clonogenic survival assays has reported DEF’s of 1.33-1.45 (concentration 30 μM-100 μM) [24] whereas benzyl isothiocyanates have reported DEF’s of 1.19 and 1.33 at concentrations of 5 μM and 10 μM respectively [25]. Certain chemotherapy drugs are often used concurrently with radiotherapy since they act by interrupting the G2-M phase of the cell cycle; the phase most sensitive to X-ray irradiation [26]. Monomethyl auristatin E (MMAE) is a synthetic antineoplastic agent, which is used as a chemotherapy agent when linked to a targeted monoclonal antibody (MAB), and acts as a potent antimitotic. Consequently it can be expected to act as an effective radio sensitizer. Clonogenic assays on PANC-1 cells treated at clinically relevant concentrations of MMAE show DEF’s of approximately 1.33 [27]. These results indicate that the use of rare earth doped titanium oxide nanoparticles shows a highly significant enhancement of the effects of radiotherapy on radio resistant cell lines such as PANC-1 when compared to chemotherapy based radio sensitizers.
FaDu in vivo xenograft study
Tumour growth rates: Human immortal cell lines grown subcutaneously as xenografts on immunocompromised mice are used extensively to evaluate the ability of cancer therapies to shrink and retard regrowth of the tumours. It has been shown that biological activity in these pre-clinical setting is reasonably well correlated with human clinical outcomes [28]. Treatment is initiated once tumours reach a certain size, typically 100-150 mm3, with tumour volumes being measured by callipers repeatedly during the treatment phase and post-treatment regrowth. Biological efficacy is measured by comparison of changes in tumour volume. Choice of end point is critical, a common and simple method is to compare the ratio of tumour volume to control (T/C) ratio at a specified time point. However this approach has limitations in that it makes no use of data acquired prior to termination of the study and bias can occur when animals are sacrificed or growth is non-linear [29]. In modelling tumour regrowth post-treatment a Gompertz curve is the most commonly used model. For theoretical reasons tumour growth should follow a Gompertz model and this has been demonstrated empirically in both animal and human tumours [30,31]. In this study tumour regrowth rates post-treatment have been ascertained by fitting a Gompertz function to the growth curves at day 16, the point at which regrowth following treatment begins.
The Gompertz function is given by:

Where y0 is the initial volume, t0 scales along the x axis and t is time, a is an asymptote and b is the tumour growth rate. Figure 2 shows tumour growth curves for FaDu xenografts over two weeks (2 cycles of 2 × 5 Gy) radiotherapy plus three weeks tumour regrowth. Radiotherapy was carried out on the days arrowed with nanoparticle injections occurring on Days 1 and 8. Group 1 gives the control (radiotherapy only) tumour response, Group 2 shows the radiotherapy plus nanoparticles tumour response, Group 3 is the control (no treatment) tumour growth curve and Group 4 is the control (nanoparticles only) tumour growth curve. Tumour volumes are the mean of the group ± the standard error. Statistical significance is calculated from Student t-test following Shapiro Wilk normality testing (Sigmaplot SPSS). FaDu xenografts grow rapidly with tumours beginning to ulcerate on mice as the tumour volume grows greater than 500 mm3, consequently the control groups 3 and 4 are plotted to 500 mm3 to maintain the same cohort number per group. The control (no treatment) and control (nanoparticles only) give the same response, within statistical error, indicating that the nanoparticles are completely inert in the absence of external beam radiotherapy. Group 1 (radiotherapy control) shows good suppression of tumour growth during treatment which is further enhanced in Group 2 by the use of nanoparticle radiotherapy enhancers. Gompertz modelling of regrowth rates from day 16 of the experiment gives a regrowth rate in Group 1 of 19.8 s-1 and Group 2 of 5.1 s-1. Therefore the regrowth rate of the tumours post-treatment is reduced by a factor of 3.8 by the use of nanoparticle enhancement concurrent with external beam radiotherapy. At Day 32 (end of experiment) the T/C ratio=0.55 with P=0.006.

Figure 2: Tumour growth curves ± SE during treatment and regrowth of FaDu oropharyngeal tumour xenografts in CD-1 nude mice. Group 1 undergo radiotherapy of 2 Gy daily on Days 1-5 and 8-12 to a total of 20 Gy. Group 2 are injected intratumourally with 20 µl of 6.25 mg.ml-1 on Days 1 and 8 and undergo radiotherapy as per Group 1. Group 3 undergo sham radiotherapy on Days 1-8 and 8-12. Group 4 are injected intratumourally with 20 µl of 6.25 mg.ml-1 on Days 1 and 8. Re-growth rates are measured by fitting a Gompertz function to Groups 1 and 2 from Day 16. Radiotherapy treatment (RT) days are indicated by arrows. Nanoparticle (NP) injection days are indicated by dashed arrows.
BrdU histology of tumour sections post-treatment
BrdU staining indicates cells which are actively replicating their DNA. Figure 3 shows typical examples of BrDu stained tumours removed from animals culled on Day 14 of the trial. There is a marked difference in proliferation between tumours treated with nanoparticle enhanced radiotherapy (Group 2 - Figure 3B) and radiotherapy alone (Group 1 - Figure 3A). There is a moderate reduction in BrdU staining between control Group (Figure 3C) and Group 1 indicating that the radiotherapy control is inducing a moderate level of cell death. No difference is observed between Groups 3 and 4 (Figure 3D) confirming that the nanoparticles are inert in the absence of external beam radiotherapy.

Figure 3: BrdU stained tumour cross-sections indicating proliferating cells from tumours removed on Day 14 of FaDu xenograft assay. Tumours are from A) Group 1-radiotherapy alone, B) Group 2-nanoparticle enhanced radiotherapy, C) Group 3-Sham radiotherapy control and D) Group 4- Nanoparticle control.
Animal weights as a proxy measurement of systemic toxicity
Figure 4 gives mean mouse weight per group throughout the in vivo trial. There is a slight decrease in animal weight during the treatment period of the trial in all groups due to the effects of anaesthetic and stresses to the animals during treatment. However, the animals soon recovered and continued to gain weight throughout the study. There is no statistically significant difference between any of the groups indicating that neither radiotherapy, nanoparticles or the combined treatment are exhibiting any systemic toxicity.

Figure 4: Mean mouse weight during trial ± SD. Group 1-radiotherapy alone (RT), Group 2- nanoparticle enhanced radiotherapy (RT+NP), Group 3-Sham radiotherapy control (Control) and Group 4-Nanoparticle control (NP). There is no statistically significant difference between the groups indicating that intratumoural injection of nanoparticles does not induce any systemic toxicity. Radiotherapy treatment (RT) days are indicated by arrows. Nanoparticle (NP) injection days are indicated by dashed arrows.
Transmission Electron Microscopy (TEM) analysis of tumours
BrdU staining indicates that nanoparticle enhanced radiotherapy results in greater tumour cell apoptosis than radiotherapy alone but does not elucidate the mechanism of tumour interaction with nanoparticles following intratumoural injection. Mathematical modelling of nanoparticle transport in solid tumours following direct injection indicates that particles on a sub-100 nm scale can diffuse throughout the tumour, interactions with cellular structures and collagen fibres being the dominant mechanism leading to nonuniform particle distribution through the tumour. The diffusion of nanoparticles is sensitive to the injection rate and surface properties of the particles but insensitive to injection volume and concentration [32]. Following interactions with cells there are a number of mechanisms of cellular uptake of nanoparticles including clathrinindependent endocytosis (≈ 90 nm), clathrin-dependent endocytosis (≈120 nm) and caveolin-dependent endocytosis (≈ 60 nm) which result in nanoparticles being transported throughout the cells in endosomes and caveosomes [33].
TEM analysis of tumours taken from mice sacrificed on Day 14 (two days following completion of treatment) show relatively uniform distribution of nanoparticles through the extracellular matrix, within which collagen fibres are clearly visible (Figure 5A) and within tumour cells, with the exception of significant preferential uptake into the Golgi apparatus of the cell (Figure 5B) (Group 2-nanoparticles plus radiotherapy). An example of the Golgi apparatus in cells removed at day 14 from control lines (Figure 5C) (Group 1-radiotherapy control) clearly shows that the contrast observed is due to the presence of nanoparticles in the tumour. Note that no agglomeration of the nanoparticles was observed. Figure 5D shows remnants of the Golgi apparatus at the end of the in vivo trial at Day 3 (Group 2- nanoparticles plus radiotherapy). Upon X-ray activation, rare earth doped titanium oxide nanoparticles generate hydroxyl free radicals which have a mean free path of 150 nm. Clearly the Golgi apparatus has been destroyed by free radicals generated by nanoparticles adjacent to cellular membranes.

Figure 5: TEM micrographs of tumour sections. A) Extracellular matrix with collagen fibres and distribution of nanoparticles. Tumour taken from Group 2-nanoparticle enhanced radiotherapy at Day 14 of in vivo trial. B) Region within tumour cell showing enhanced passive uptake into the Golgi apparatus of nanoparticles. Tumour taken from Group 2-nanoparticle enhanced radiotherapy at Day 14 of in vivo trial. C) Region within tumour cell showing Golgi apparatus. Tumour taken from Group 1-radiotherapy at Day 14 of in vivo trial and D) Region within tumour cell showing destruction of the Golgi apparatus and residual nanoparticles. Tumour taken from Group 2-nanoparticle enhanced radiotherapy at Day 32 of in vivo trial.
Targeting of the Golgi apparatus as an anticancer treatment
Nanoparticles will undergo passive uptake into the Golgi apparatus following endocytosis in accordance with well understood internalisation pathways [33]. Fragmentation of the Golgi apparatus is an early apoptotic event and induction of cell death is a critical event defining tumour response to treatment [34]. Evasion of programmed cell death is one of the distinguishing characteristics of cancer and the exploration of alternative pathways to apoptosis are currently being explored by routes involving the endoplasmic reticulum, lysosomes and the Golgi apparatus [35,36]. Indeed, destruction of the Golgi apparatus and consequent cell apoptosis is one of the mechanisms of neurodegeneration occurring during the progression of Alzheimer’s disease [37]. In addition to the Golgi’s well known importance in processing and sorting of lipids and proteins recent work suggests additional functions related to initiating pathways to relieve cellular stress and, if appropriate, triggering apoptosis [38]. Golgi fragmentation and destruction is often observed subject to oxidative or pharmacological stress; signalling pathways act to correct the stress if possible and, if impossible, trigger apoptosis [39]. The use of rare earth doped titanium oxide nanoparticles to enhance oxidative stress on cancer cells generally, and in particularly to initiate complete fragmentation of the Golgi apparatus via X-ray induced free radical damage, appear to be a useful route to radiotherapy enhanced treatment of solid tumours and improved cancer therapy.
Conclusions
Rare earth oxide doped titanium oxide nanoparticles generate hydroxyl free radicals by water splitting when excited by incident X-rays or secondary electrons generated by X-ray Compton scattering. In vitro and in vivo studies demonstrate that this increases the effectiveness of external beam radiotherapy in the treatment of solid tumours without any increased systemic toxicity. In vitro studies utilising PANC-1 pancreatic cancer clonogenic assays indicate a dose enhancement function of 1.9 at clinically relevant dosing. In vivo assays utilising FaDu oropharyngeal cancer xenografts show a marked reduction in tumour regrowth rates above those achievable by conventional radiotherapy. This is shown to be a result of increased levels of cell apoptosis, which is as a result of passive accumulation and destruction of the Golgi apparatus by X-ray induced photo activity during the application of radiotherapy.
Acknowledgements
The authors would like to thank Dr Saskia Bakker and Dr Ian Hands-Portman of the University of Warwick, UK, for TEM imaging and Dr Greg Tudor, Dr Sarah Hoyle and Ms Emma (Bowden) Playle of Epistem Ltd, Manchester, UK, for in vitro and in vivo studies.
References
- Delaney G, S Jacob, Featherstone C, Barton M (2005) The Role of Radiotherapy in Cancer Treatment. Cancer 104: 1129-1137.
- Round CE, Williams MV, Mee T, Kirkby NF, Cooper T, et al. (2013) Radiotherapy Demand and Activity in England (2006-2020). Clin Oncol 25: 522-530.
- Barton MB, Gebski V, Manderson C, Langlands AO (1995) Radiotherapy: Are we Getting Value for Money. Clin Oncol 7: 287-292.
- Morgan G, Ward R, Burton M (2004) The Contribution of Cytotoxic Chemotherapy to 5 Year Survival in Adult Malignancies. Clin Oncol 16: 549-560.
- Wagstaff A (2005) Radiotherapy Report Sets New Target for Europe. Cancer World: 32-36.
- Plonquin NP, Dunscombe PB (2008) The Costs of Radiotherapy. Radiother Oncol 86: 217-223.
- Podgorsak EB (2005) Radiation Oncology Physics: A Handbook for Teachers and Students, IAEA
- Brunner TB, Nestle U, Grosu AL, Partridge M (2015) SBRT in Pancreatic Cancer: What is Therapeutic Window? Radiother Oncol 114: 109-116.
- Chaudary N, Hill RP (2007) Hypoxia and Metastasis. Clin Cancer Res 13: 1947-1949.
- Vaupel P (2008) Hypoxia and Aggressive Tumour Phenotype: Implications for Therapy and Prognosis. The Oncologist 13 Suppl. 3: 21-26.
- Hainfeld JF, Slatkin DN, Smilowitz (2004) The use of gold nanoparticles to enhance radiotherapy in mice, Phys Med Biol 49: N309-N315.
- Chang MY, Shiau AL, Chen YH, Chang CJ, Chen HHW, et al. (2008) Increased apoptotic potential and dose-enhancing effect of gold nanoparticles in combination with single dose clinical electron beams on tumor-bearing mice. Cancer Sci 99: 1479-1484.
- Marill J, Anesary NM, Zhang P, Vivet S, Borghi E, et al. (2014) Hafnium Oxide Nanoparticles: Toward an in vitro predictive biological effect? Radiat Oncol 9: 150-161.
- Hext P, Tomenson JA and Thompson P (2005) Titanium Dioxide: Inhalation Toxicology and Epidemiology. Ann Occup Hyg 49: 461-472.
- Bingham S, Daoud WA (2011) Recent advances in making nano-sized TiO2 visible-light active through rare-earth metal doping. J of Mater Chem 21:2041-2050.
- Dvoranová D, Bresová V, Mazúr M, Malati MA (2003) Investigations of metal doped titanium dioxide photocatalysts. Appl Catal B Environ 37: 91-105.
- Townley HE, Kim J, Dobson PJ (2012) In vivo demonstration of enhanced radiotherapy using rare earth doped titania nanoparticles. Nanoscale 4: 5043-5050.
- Tang L, Yang X, Yin Q, Cai K, Wang H, et al. (2014) Investigating the optimal size of anticancer nanomedicine. PNAS 111: 15344-15349.
- Franken NAP, Rodermond HM, Stap J, Haveman J, van Bree C (2005) Clonogenic assay of cell in vitro. Nature Protocols 1: 2315-2319.
- Gradiz R, Silva HC, Carvalho L, Botelho MF, Mota-Pinto A (2016) MIA PaCa-2 and PANC-1- pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Scientific Reports 6: 21648.
- Jung J (2014) Human Tumour Xenograft Models for Preclinical Assessment of Anticancer Drug Development. Toxicol Res 30: 1-5.
- Rieckmann T, Tribius S, Grob TJ, Meyer F, Busch TJ, et al. (2013) HNSCC cell lines positive for HPV and p16 possess higher cellular radio sensitivity due to an impaired DSB repair capacity. Radiother Oncol 107: 242-246.
- Mohamed NA, Cricco GP, Cocca CM, Rivera MS, Bergoc RM, et al. (2012) PANC-1 cells proliferative response to ionizing radiation is related to GSK-3β phosphorylation. Biochem Cell Biol 90: 779-790.
- Fasih A, Elbaz HA, Hüttemann M, Konski AA, Zielske SP (2014) Radio sensitization of pancreatic cancer cells by metformin through the AMPK pathway. Radiat Res 182: 50-59.
- O’Hara M, Kimura S, Tanaka A, Ohnishi K, Okayasu R, et al. (2011) Benzyl isothiocyanate sensitizes human pancreatic cancer cells to radiation by inducing apoptosis. Int J Mol Med 28: 1043-1047.
- Maity A, McKenna G, Muschel RJ (1994) The molecular basis for cell cycle delays following ionising radiation: a review. Radiother Oncol 31: 1-13.
- Buckel L, Savariar EN, Crisp JL, Jones KA, Hicks AM, et al. (2015) Tumor Radio sensitization by Monomethyl Auristatin E: Mechanism of Action and Targeted Delivery. Cancer Res 75: 1376-1387.
- Kelland LR (2004) “Of mice and men”: values and liabilities of the athymic nude mouse model in anticancer drug development. Eur J Cancer 40: 827-836.
- Hather G, Liu R, Bandi S, Mettetal J, Manfredi M, et al. (2014) Growth rate analysis and efficient experimental design for tumour xenograft studies. Cancer Inform 13: 65-72.
- Heitan DF (1991) Generalised Norton-Simon modals of tumour growth. Statis Med10: 1075-1088
- Laird AK (1964) Dynamics of tumour growth. Br J Cancer 18: 490-502.
- Du S, Ma R, Salloum M, Zhu L (2010) Multiscale study of nanoparticle transport and deposition in tissues during an injection process. Med Biol Eng Comput 48: 853-863
- Yameen B, Choi WI, Vilos C, Swami A, Shi J, et al. (2014) Insight into nanoparticle cellular uptake and intracellular targeting. J Contr Rele 190: 485-499.
- Mukherjee S, Chiu R, Leung SM, Shields D (2007) Fragmentation of the Golgi apparatus: an early apoptotic event independent of the cytoskeleton. Traffic 8: 369-378.
- Levine B, Yuan J (2005) Autophagy in cell death: an innocent convict? J Clin Invest 115: 2679-2688.
- Wlodkowic D, Skommer J, McGuinness D, Hiller C and Darzynkiewicz Z (2009) ER-Golgi network- a future target for anti-cancer therapy. Leuk Res 33: 1440-1447.
- Joshi G, Bekier ME, Wang Y (2015) Golgi fragmentation in Alzheimer's disease. Frontiers in Neuroscience 9: 340.
- Hicks SW, Machamer CE (2005) Golgi structure in stress sensing and apoptosis. Biochim BiophysActa 1744: 406-414.
- Machamer CE (2005) The Golgi complex in stress and death. Front Neurosci 9: 421.