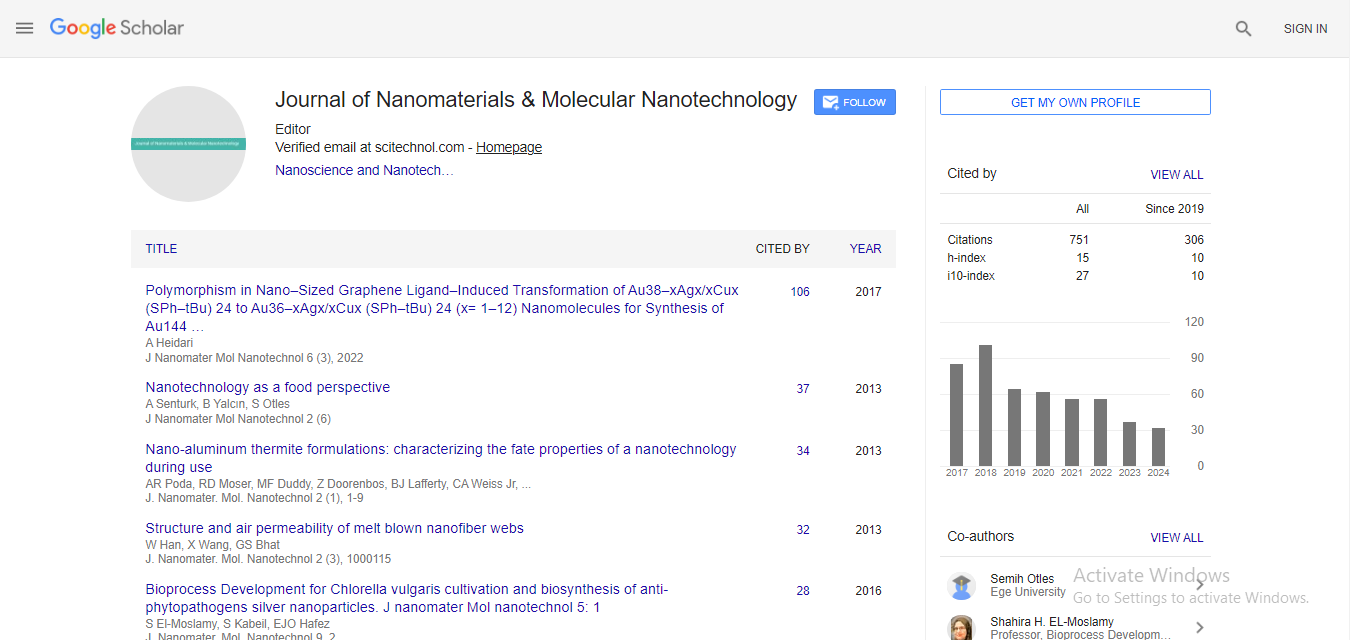
Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Research Article, J Nanomater Mol Nanotechnol Vol: 5 Issue: 6
A Novel Carbon Paste Electrode Based on Ion-Imprinted Polymer for Determination of Iridium
| Huiping Bai1,2, Chunqiong Wang3, Caiyun Xiong1, Miao Guo1,Longchun Bian1 and Qiue Cao1* | |
| 1School of Chemical Science and Technology; Yunnan University, Kunming 650091, Chin | |
| 2Yunnan Key Laboratory of Micro/nano Materials & Technology; Yunnan University, Kunming 650091, China | |
| 3Yunnan Tobacco Quality Supervision and Test Station, Kunming 650106, China | |
| Corresponding author : Huiping Bai
Qiue Cao, School of Chemical Science and Technology; Yunnan University, Kunming 650091, China E-mail: baihuiping@ynu.edu.cn; qecao@ynu.edu.cn |
|
| Received: July 18, 2016 Accepted: August 05, 2016 Published: August 11, 2016 | |
| Citation: Huiping Bai, Chunqiong Wang, Caiyun Xiong, Miao Guo, Longchun Bian, et al. (2016) A Novel Carbon Paste Electrode Based on Ion-Imprinted Polymer for Determination of Iridium. J Nanomater Mol Nanotechnol 5:6. doi:10.4172/22324-8777.1000199 |
Abstract
A Novel Carbon Paste Electrode Based on Ion-Imprinted Polymer for Determination of Iridium
To develop a convenient method for sensitive and selective determination of iridium in complicated matrices, a carbon paste electrode based on iridium ion imprinted polymer (IIP) was fabricated. Iridium ion selective cavities were created in the 2-(allylthiol) nicotinic acid based cross-linked polymer. In order to fabricate the sensor, carbon particles and polymer powder were mixed with melted n-eicosane. An explicit difference in the response was observed between the electrodes modified with ion imprinted polymer (IIP) and non imprinted polymer (NIP), indicating proper performance of the recognition sites of the IIP. Various factors, known to affect the response behavior of selective electrode, were investigated and optimized. The resulting sensor named as Ir(III) ion imprinted polymer carbon paste electrode (Ir(III)-IIP/CPE) exhibits high response sensitivity to Ir(III) in acetate buffer (pH 3.6). The calibration graph is linear in the range of 2.85×10-8∼2.31×10-5 mol L-1 with the detection limit of 7.84×10-9 mol L-1(S/N). The electrode showed high selectivity for iridium in the presence of common potential interferers was found to show satisfactory results, it was successfully applied to the determination of iridium in real samples.
Keywords: Iridium (III); Carbon paste electrode; Ion imprinted polymer; Environmental samples
Keywords |
|
| Iridium (III); Carbon paste electrode; Ion imprinted polymer; Environmental samples | |
Introduction |
|
| The problem relevant to the presence of platinum group metals (PGMs) in the environment, normally at ultratrace level concentration, and specifically in particulate matter (PM<10 μm) owing to vehicle emissions [1-3], considered a new class of pollutants, and has been attracting researchers increasing interest and attention. In fact, the use of autocatalytic converters, containing platinum group metals (PGMs), allows obtaining the decrease of pollutants in exhaust gases from motor vehicles, like lead, carbon monoxide, nitrogen oxides and unburned hydrocarbon. But, contemporaneously, it is the cause of a widespread distribution of fine particulate matter and dust originated from deterioration or abrasion of the bulk catalysts [4,5]. | |
| In the last decade, and always more frequently, iridium is employed as alloying (10-20% w/w) with platinum, palladium and rhodium in the manufacture of autocatalytic converters in order to withstand high temperature and high wear [1]. However, these elements, widely used lead to the increasing levels of contamination in environmental matrices, especially in soils, plants and road sediments [6] as well as in airborne particles [7], and further imperil human health together with increasing the environmental risk. Thus, it is necessary to develop a convenient and reliable method to determine iridium in environmental matrices. | |
| So far, some analytical techniques including spectrophotometric methods [8,9], atomic absorption spectrometric [10,11], and flow injection analysis [12], have been applied to deter iridium of spent catalyst. These techniques are the most commonly used tools by the virtue of their wide applications and high efficiency, but they are expensive and not suitable for real-time detection. In comparison, electrochemical methods are the most favorable techniques for the determination of metal ions because of their high sensitivity in addition to low costs, ease of operation and portability. Orecchio [13] reported an extremely sensitive stripping voltammetric method for ultra-trace determination of iridium (III). The method is based on the interfacial accumulation of the iridium(III)-CTAB (Cetyl trimethylammonium bromide) complex onto the glassy carbon electrode, followed by the catalytic reduction of the adsorbe d complex in the presence of bromate. The limit of detection was of the order of 2-3 ng L-1. | |
| Carbon paste electrodes (CPEs) have attracted attention as ion selective electrodes mainly due to their advantages over membrane electrodes such as renewability, stable response, low ohmic resistance, no need for internal solution [14-16]. Ion imprinted polymer (IIP) have high selectivity and affinity similar to those of antibodies, which is very suitable for artificial recognition receptors in chemical sensors. Recently, we have reported several papers describing the application of MIP/IIP (MIP-Molecularly Imprinted Polymers correction for Molecularly/Ion Imprinted Polymers.) as a recognition element of the electrochemical sensors for determination of different kinds of molecular and ion analytes [17-19]. Due to the high selectivity of these materials, the electrode containing imprinted materials tends to show selective behaviors. Besides, this modifying agent can pre-concentrate the analyte in the electrode due to the high adsorption capacity of these materials. The obtained interesting results from the previous works provoked us to design a novel Ir(III) electrochemical sensor, using ion imprinted polymers. IIP has a high selectivity even more than organic chelating agents. It provides high adsorption capacities comparable with that of clays and zeolite, providing high sensitivity and lower detection limit. Also, IIP materials usually show a high stability and durability against harsh chemical environments [20-22]. In other words, electrodes modified with ion imprinted polymers bring together the advantages of different kinds of modifying agents in a single modified electrode. Moreover, both IIP and carbon paste are cheap materials and thus a carbon paste electrode modified with IIP can provide an efficient and cheap sensor for Ir(III) determination (Supplementary file 1). | |
| In this study, combining the advantages of ion imprinted technique and CPE detection, an IIP/CPE has been developed for the determination of iridium ions in environment sample (Supplementary file 2). The experimental parameters together with the analytical application and selectivity behavior of the IIP/CPE were investigated in detail. To our knowledge, there are no previous reports on the determination of Ir(III) using ion imprinted polymer as a recognition element in the carbon paste electrode composition and its using as a amperometric sensor for trace level Ir(III) determination. | |
Experimental |
|
| Apparatus and Instrumentation | |
| Cyclic voltammetric (CV) and Amperometric measurements were carried out with a CHI 660 D electrochemical workstation (Shanghai CH Instruments Co., China) and 797 VA Computrace (Metrohm, Switzerland). A three electrode cell (10 mL) with the carbon paste electrodes modified with IIP or NIP were used as a working electrode, a saturated calomel electrode as reference electrode and a platinum wire electrode as counter electrode was used. All potentials were measured and reported V vs revised versus. | |
| Reagents and Chemicals | |
| H2PtCl6, PdCl2, RhCl3·3H2O, IrCl3·3H2O, RuCl3 and OsCl3 were obtained from Kunming Institute of Precious Metals (Yunnan, China). 2-acetamidoacrylic acid (AAA), 2-(allylthiol) nicotinic acid (ANA), Methacrylic acid (MAA), and acrylamide (AM) were purchased from Sigma Aldrich (St. Loius, MO, USA). Ethylene glycol dimethacrylate (EGDMA) was purchased from Suzhou Anli Chemical Factory (Jiangsu, China) and distilled under vacuum to remove the stabilizers prior to use. Azobisisobutyronitrile (AIBN) was purchased from Shanghai Reagent Factory (Shanghai, China) and purified by recrystallization from ethanol before used. The other reagents and solvents were purchased from Tianjin Kemiou Chemical Reagent Co., Ltd. (Tianjin, China). | |
| Preparation of sample solution | |
| Treatment of the leaves sample solutions: About 100 g of leaves were collected from the cinnamomum pedunculatum in 121 street of Kunming (Yunnan, China). The leaves were firstly washed with detergent, tap water and de-ionized water successively, and then dried at room temperature for 24 h followed by drying at 90oC for 4 h. After that, 5.0000 g of the pulverized leaves was immersed with 10 mL of nitric acid for 12 h in a conical flask covered with a short neck funnel, and then heated at 140oC until the volume of solution was less than 1 mL. 5 mL of aqua regia was added. The mixture was heated at 160oC to near dryness and the residue was dissolved with 5 mL of de-ionized water by boiling for 10 min. After cooling to room temperature, the solution was filtered, and the filtrate was diluted to 25 mL with de-ionized water. | |
| Preparation of the catalyst sample solutions: After the catalyst materials were ground to pass through a 200 mesh size sieve to facilitate sample dissolution, 25.0 mg of samples was weighed accurately into a Teflon high-pressure microwave acid digestion bomb and 5.0 mL of concentrated nitric acid, 4.0 mL of hydrochloric acid and 10.0 mL of 30% hydrogen peroxide were added. The bomb was sealed tightly and then positioned on the carousel of the microwave oven. The system was operated at 600 W and 350 psi with a heating program as 120oC (5 min, hold 2 min), 180°C (6 min, hold 10 min). The digest was evaporated to near dryness. The residue was dissolved with 2 mL of 2.0 mol L-1 HCl and then dilluted to 25 mL with de-ionized water. | |
| Synthesis of Ir(III)-IIP | |
| Ir(III)-IIP was prepared by precipitation polymerization using IrCl3·3H2O as template, ANA as functional monomer, EGDMA as crosslinker, AIBN as initiator and methanol as porogen. The typical procedure for preparing IIP was as follows: IrCl3·3H2O (0.05 mmol), ANA (0.1 mmol) and EGDMA (2 mmol) were dissolved in 10 mL of Methanol. Then, 9 mg of the initiator (AIBN) was added in the previous solution and purged with N2 gas for 10 min. The polymerization was carried out in a water bath at 80°C for 24 h. The obtained polymer particles were firstly washed with ethanol/ acetic acid (v/v=9:1) solution. Finally, the particles were washed with ethanol and dried at 60°C. The NIP was prepared, using the same protocol in the absence of Ir(III). The prepared IIP and NIP were used for carbon paste electrode fabrication. | |
| Preparation of carbon paste electrode | |
| For construction of the carbon paste electrode (IIP/CPE), 0.02 g graphite was homogenized in a mortar with 0.01 g of powdered IIP for 10 min. Subsequently, n-eicosane, 18 μL was dropped in a dish in a water bath, and heated at 45-50oC. The graphite/IIP blend was then added to the n-eicosane and mixed with a stainless steel spatula. The final paste was used to fill a hole (2.00 mm in diameter and 3.00 mm in depth) at the end of an electrode body, previously heated at 45oC. After cooling at room temperature, the excess of solidified material was removed with the aid of a sheet of paper sheet. The electrode can be reused after each experiment by moving the electrode surface on a paper sheet in order to rub out a thin layer of the electrode surface. | |
| Electroanalytical measurements | |
| The electrochemical characteristics of the modified electrode were characterized by Cyclic Voltammetric and Amperometric measurements. Cyclic voltammetric measurements were carried out in 10 mL of 0.2 mol L-1 acetate buffer solution (pH 3.6) at room temperature from -0.8 V to +0.8 V with a scan rate of 100 mV s-1. The amperometric detection was based on the change in the current response (Δi) before and after different concentrations of Ir(III) was added in acetate buffer solution and the potential for the determination of Ir(III) was -0.9 V. | |
Results and discussion |
|
| Optimization of the conditions for Ir(III)-IIP | |
| Selection of functional monomer: Since the principle of the ion imprinting technique lies in the preservation of the prepolymerized host-guest structure into a polymer matrix, it is crucial that the template and monomer forms a stable host-guest complex in the prepolymerization system [20]. In this study, the interaction between the template ion Ir(III) and four functional monomers (ANA, MAA, AM and AAA) in polymerization period was investigated by UV spectrophotometry. | |
| The UV-spectra of Ir(III) in the absence or presence of different monomers point out that the additions of ANA, MAA, AM and AAA all can result in red shift together with hypochromic effect of the absorption peak of Ir(III) located at about 215 nm, and the most obvious red shift and hypochromic effect is obtained by adding ANA (Figure 1A). These results indicate that Ir(III) can bind to each functional monomers tested and the interaction between Ir(III) and ANA is the strongest compared with the interaction between Ir (III) and the other monomers. | |
| Figure 1: UV spectra of Ir(III) and its mixture with (A) different functional monomer and (B) different concentration of ANA in methanol. The concentration of Ir3+ is 0.5 mmol L-1, the concentration of functional monomer in figure 1A is 2.0 mmol L-1 and concentration of ANA for line 1 to line 8 in figure 1B is: 0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0 and 3.5 mmol L-1. The reference is corresponding methanol solution containing the same concentration of functional monomer | |
| Assuming Ir(III) (P) and ANA (M) in ethanol formed the composition
ratio is 1: n complexes (PMn), in accordance with the literature
[21] has equations: |
|
| Optimization of the cross-linking agent: The quantity of imprinting recognition sites in ion imprinted polymers is a direct function of the degree of cross-linking in the polymerization. In order to optimize cross-linking for the preparation of IIP, molar ratio of Ir(III)/ANA was fixed at 1:2, and imprinted polymer of different molar ratios of Ir(III)/EGDMA (1:30, 1:40, 1:50, 1:60, 1:70, 1:80) were prepared and used for carbon paste electrode fabrication. The performance of IIP/CPE was investigated through the current response of the IIP/CPE to 1.2 μmol L-1 Ir(III) in the acetic acid buffer solution. The results point out that the IIP/CPE prepared with 1:40 (molar ratio) shows the maximum response sensitivity to Ir(III). And the other sensors prepared with more or less molar ratio of 1:40 possess relatively weak current response. This may be due to the fact that if the amount of crosslinking agent was too small, the polymer formation of functional monomer and template molecules cannot be fixed, resulting in fewer imprinted cavities. While the excess crosslinking agent would lead to the specific surface area of polymer reduction, and imprinted locus were embedded in polymer, which made the mass transfer of imprinted ions in the polymerization system were blocked, so that the adsorption capacity of polymer for rhodium ions recede, recognition ability weakens [22]. Therefore, the molar ratio of Ir(III)/EGDMA for synthesis of Ir(III)-IIP was selected as 1:40. | |
| Optimization of the composition of IIP/CPE | |
| In order to find the best composition for the IIP/CPE, the amounts of IIP was changed and the obtained responses were used for conclusion. The IIP/CPE was prepared with 0.02 g carbon and different amounts of IIP. The fabricated electrode, in each case, was inserted in acetate buffer (pH 3.6) containing 1.2 μmol L-1 Ir(III) that the maximum response for the prepared sensor appears in the IIP amount of 0.01 g. The higher the amounts of IIP are the sensor response increases due to providing more recognition sites on the electrode surface. However, increase the IIP above a threshold amount, leads to a decrease in the response of the prepared sensor; most probably due to decrease in the conductivity of the electrode surface, this conclusion is consistent with the reported literature [23]. | |
| Optimization of the determination conditions | |
| In order to improve the sensitivity of the sensor, determination conditions liked the pH of electrolyte solution and applied potential were optimized. The results are shown in Figure 2. As can be seen, stabilizing the pH is crucial task in this work. The maximum current response of IIP/CPE is obtained in acetate buffer with pH of 3.6, and all of the other buffers or acetate buffer with pH more or less than 3.6 produce a decrease in the current. Therefore, pH 3.6 acetic acid buffer solution was used for the determination of Ir(III) to achieve higher sensitivity. | |
| Figure 2: The current response of Ir(III)-IIP/CPE to 1.2 �?µmol L-1 Ir(III) in (a) citric acid, (b) acetic acid, and (c) phosphate buffer solution. | |
| The effect of applied potential on the sensor response was studied in acetate buffer (pH 3.6) containing 1.2 μmol L-1 Ir(III). The response current of the IIP/CPE increases with negative shift of the potential in the range of -0.6 to -1.1 V. To avoid interference at high negative applied potential, a potential of -0.9 V (vs. SCE) was selected as the applied potential for amperometric measurements. | |
| Characterization of the Ir(III)-IIP | |
| The resulting Ir(III)-IIP was characterized by Fourier transform infrared spectroscopy (FT-IR), and scanning electron microscopy (SEM). The surface morphology of the Ir(III)-IIP was shown by SEM as depicted in Figure 3. It can be seen from the SEM images that the Ir(III) imprinted particles were uniform and spherical with a diameter of about 1 μm. These uniform-sized micron-particles can be well dispersed in the graphite of CPE, which could induce more binding sites available in the surface of CPE. | |
| Figure 3: SEM photographs of Ir(III) ion-imprinted polymer. | |
| The surface area, pore volume and average pore radius of the polymers were determined by BET nitrogen adsorption analysis after the sample was heated at 150oC under high vacuum for 30 min to be 9.42 m2 g-1, 0.0246 cm3 g-1 and 13.6 nm for IIP, which are higher than that of NIP (i.e. 7.51 m2 g-1, 0.0193 cm3 g-1 and 10.17 nm, respectively),confirming that the pore properties of IIP are enhanced by the imprinting process. | |
| Figure 4 is the FT-IR spectra of the un-leached (a), leached(b) Ir(III)-IIP, and NIP (c) recorded by KBr pellet method. Visible, the infrared spectrum of leached IIP and NIP polymer is very similar, the number and location of absorption peak has not changed, indicating that the two kinds of polymer possess a similar chemical structure and the leaching process does not affect the polymeric network. Compared with the leached polymer (b), un-leached polymer (a) is almost no change in the peak of carbonyl groups (vC=O, 1732 cm-1) and hydroxyl (vO-H, 3448 cm-1), but the obviously shift of absorption peaks in 1632 cm-1, 1458 cm-1, 1267 cm-1 and 927 cm-1 are observed. One reason may be the stretching vibration peak of pyridine ring skeleton in 1632 cm-1 and 1458 cm-1 respectively displacement to the 1643 cm-1 and 1467 cm-1. Another reason are C-S stretching vibration peak by the displacement of 1267 cm-1 to 1260 cm-1, and the C-H bending vibration peak on the pyridine ring in 927 cm-1 displacement to 950 cm-1. The change of the position of the characteristic peak and the increase of the number of absorption peaks are the evidences for the formation of complexes [24]. According to the change of the characteristic peak, ANA can bind with Ir(III) using nitrogen atom and sulfur atom as donor atoms. After the formation of the complex, the absorption peak of the pyridine ring shifted towards high frequency, this is caused by the vibration disruption of Ir(III) combine with nitrogen and sulfur atoms of ligand [28]. | |
| Figure 4: IR spectra of imprinted polymers before (a), after (b) template removal and non-imprinted polymers (c). | |
| Electrochemical characterization of the IIP/CPE | |
| The electrochemical property of the resulting IIP/CPE, NIP/CPE and CPE was investigated by cyclic voltammetry in 10 mL of acetic acid buffer solution (pH 3.6) without Ir(III) (Figure 5A) or with 6.0 μmol L-1 Ir(III) (Figure 5B). It can be seen from Figure 5A that no redox peaks were observed for CPE (a), NIP/CPE (b) and IIP/CPE (c) in acetic acid buffer solution without Ir(III), suggesting that they were not oxidized or reduced in the potential range from -0.8 to 0.8 V. While the solution containing 6.0 μmol L-1 Ir(III) (Figure 5B), an obvious anodic stripping voltammetric peaks was observed for IIP/ CPE (curve c), no anodic peak or only a small signal was observed for CPE and NIP/CPE (curve a and b). These results demonstrate that Ir(III) was reduced and enriched only in the IIP/CPE, which might be attributed to the specific recognition sites existing in the IIP. The imprinted sites not only have recognition characteristics, but also play a role of enrichment, which could enhance the amperometric response to Ir(III). | |
| Figure 5: Cyclic voltammograms of bare CPE (a), NIP-CPE (b) and Ir(III)-IIP/CPE (c) in acetate buffer (pH 3.6) (A) without or (B) containing 6.0 �?µmol L-1Ir(III). | |
| Amperometric i-t curve | |
| The affinity of the resulting Ir(III)-IIP/CPE, NIP/CPE and bare CPE to Ir(III) in the concentration range over 0.0285∼43.6 μmol L-1 were investigated by amperometric i-t curve at the potential of -0.9V. As shown in Figure 6, the amperometric currents of bare CPE is almost not varied with the concentration of Ir(III), but the amperometric currents of the IIP/CPE increased with the increment of Ir(III) concentration. Noticeably, the sensitivity of IIP/CPE is significantly greater than that of the NIP/CPE, indicating that the specific recognition sites of IIP can enhance amperometric response to Ir(III). The mechanism of facilitated transport and lack of specific binding sites can be applied to explain the great difference between the amperometric current of IIP/CPE and NIP/CPE. | |
| Figure 6: Typical current response i-t curve of Ir(III)-IIP/CPE, bare CPE and NIP-CPE with increasing addition of Ir(III) in acetate buffer (pH 3.6) at the applied potential of -0.9 V. Concentration (�?µmol L-1) of Ir(III): a: 0.0285, b:0.314, c: 0.599, d: 0.884, e: 1.45, f: 2.59, g: 4.87, h: 7.15, i: 9.43, j: 11.7,k: 14.0, l: 16.3, m: 19.7, n: 23.1, o: 32.2, p: 37.9, q: 43.6. Inset figure is the calibration curve. | |
| The current response of IIP/CPE increased with increasing of the concentration of Ir(III), and the relationship between the response currents and concentrations of Ir(III) shown as inset in Figure 6. It can be seen that the calibration curve for the electrochemical determination of Ir(III) is composed of two distinct straight lines, suggesting that there are two types of binding sites, which can be named as high affinity binding sites and low affinity binding sites, existed in the Ir(III)-IIP/CPE. This result is consistent with many studies of imprinted polymers [26]. The linear regression equations in the Ir(III) concentration range over 2.85×10-8~1.45×10-6 mol L-1 and 1.45×10-6~2.31×10-5 mol L-1 are obtained as ΔI=30.86c (μmol L-1)- 0.77(r=0.9981) and I=67.38c (μmol L-1)-51.53(r=0.999), respectively. The limit of detection of the sensor, which is defined as the lowest concentration of Ir(III) producing a peak current 3 times higher than the standard deviation of 9 blank measurements, is found to be 7.84 nmol L-1. | |
| Selectivity, repeatability and stability of IIP/CPE | |
| The selectivity of the resulting IIP/CPE was evaluated by testing the response current of this sensor to 0.50 μmol L-1 Ir and different concentrations of other metal ions. As it is clear in Table 1, the concentrations of Cu2+, Fe3+, Zn2+, Al3+, Mg2+, Ca2+, Ba2+ are 500 times as much as that of Ir(III). Ag (I), Pb (II), Si (IV) and Ni (II) are 200 times. Pt (IV), Rh (III), Ru (III), Pd (II), Os (IV) and Au (III) are 18 times. It must be noticed that the signals observed for these cations in the electrode are in the potential ranges that are far weaker than Ir(III), approximately meaning that these ions have no interference in the determination of Ir(III) even at higher concentrations of the described interferer ions. Therefore, the imprinted polymer only has a high selectivity to iridium ions. | |
| Table 1: The response current (ΔI) of IIP/CPE to different metal ions | |
| Reproducibility and stability of the sensor was also investigated. For five successive determinations of 1.2 μmol L-1 Ir(III) at the same modified electrode, the relative standard deviation (R.S.D.) was 4.8%. Fabrication reproducibility was estimated with five different sensors, which were constructed by the same procedure, R.S.D. was 6.4%. The results showed that the proposed IIP/CPE had good repeatability and stability. | |
| Real sample analysis | |
| Two real samples, including an automotive catalyst material and a plant sample, were analyzed by the resulting Ir(III)-IIP/CPE to evaluate the precision and accuracy of the proposed electrochemical method (Table 2), which was obtained via the standard addition method, are in the range of 98.7∼101.7%. To further verify the accuracy of the quantitative results obtained by Ir (III)-IIP/CPE, the concentrations of iridium in sample solutions were also quantified by AAS (Atomic Absorption Spectroscopy) method. Iridium does not be determined in plant sample solution by AAS and the concentration of iridium in catalyst sample solution is 2.30×10-6 mol L-1. It can be seen that the quantitative results of iridium obtained by Ir(III)-IIP/ CPE are in agreement with that obtained by AAS, suggesting that the resulting Ir(III)-IIP/CPE may be a feasible tool for the determination of Ir(III) in the complicated samples. | |
| Table 2: Determination of Ir(III) in real samples by Ir(III)-IIP/CPE (n=5). | |
Conclusions |
|
| The results of this study show that the amperometric method using modified carbon paste electrode provides an attractive alternative for the determination of Ir(III). Application of IIP as a novel modifying agent in the carbon paste electrode made it very selective for iridium determination in the presence of common potential interfering agents. And the IIP inside of carbon paste electrode can not only act as the selective inducing agent, but also play the role of a pre-concentrator. As a result, the developed sensor exhibits good selectivity and high sensitivity. The electrochemical method proposed based on the resulting IIP/CPE can be used for the direct determination of iridium at trace levels in complicated matrices. | |
Acknowledgments |
|
| This work was financially supported by the Natural Science Foundation of China (No.21465025) and Academician Free Inquiry Project of Yunnan Province (No. 2015HA015). | |
References |
|
|
|